

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Original Article
Volume 4, Number 2, April 2012, pages 95-101
Nephrotoxin-Induced Renal Cell Injury Involving Biochemical Alterations and Its Prevention With Antioxidant
Andrew I. Fishmana, Bobby Alexandera, Majid Eshghia, Muhammad Choudhurya, Sensuke Konnoa, b
aDepartment of Urology, New York Medical College, Valhalla, New York, USA
bCorresponding author: Sensuke Konno, Department of Urology, New York Medical College, Munger Pavilion 4th Floor, Valhalla, NY 10595, USA
Manuscript accepted for publication February 16, 2012
Short title: Biochemical Alterations With Nephrotoxin
doi: https://doi.org/10.4021/jocmr833w
| Abstract | ▴Top |
Background: Although nephrotoxic agents or nephrotoxins are known to induce acute renal cell injury, their cytotoxic action is not fully elucidated. It is thus crucial to explore such a cytotoxic mechanism and the increasing volume of reports indicated a significant involvement of oxidative stress. To test this possibility, we investigated if a nephrotoxin would exert oxidative stress, leading to renal cell injury accompanied by certain biochemical alterations. We also examined if specific antioxidant might help prevent such oxidative cell injury. These studies may then help establish a prophylactic or preventive modality for renal cell injury induced by nephrotoxins.
Methods: As glycerol has been commonly used for studying acute renal failure in animals, whether it would induce cellular injury was tested in renal proximal tubular OK cells in vitro. Cells were exposed to the varying concentrations of glycerol and cell number/viability was determined in 24 hours. Severity of oxidative stress was assessed by lipid peroxidation assay. Possible effects of glycerol on biochemical parameters were also examined on glyoxalase I activity and heat shock protein 90 using spectrophotometric (enzymatic) assay and Western blot analysis.
Results: Glycerol (2.5%) was highly cytotoxic to OK cells, inducing 95% cell death in 24 hours. Lipid peroxidation assay indicated that nearly 3-fold greater oxidative stress was exerted by this glycerol. Concurrently, glyoxalase I activity was drastically lost by 75% and heat shock protein 90 was partially degraded following glycerol exposure. However, N-acetylcysteine, a potent glutathione-based antioxidant, was capable of almost completely preventing the glycerol-mediated adverse outcomes, such as cell death, glyoxalase I inactivation, and heat shock protein 90 degradation.
Conclusions: Glycerol is cytotoxic, capable of inducing specific biochemical alterations such as inactivation of glyoxalase I and degradation of heat shock protein 90, which may reflect a breakdown of the cellular detoxification and defense systems, leading ultimately to OK cell death. Nevertheless, as N-acetylcysteine can provide full cytoprotection against such glycerol toxicity, it could be considered a prophylactic modality for nephrotoxin-induced oxidative renal cell injury and death.
Keywords: Glycerol; Glyoxalase I; Heat shock protein; N-acetylcysteine; Renal cell injury
| Introduction | ▴Top |
Various nephrotoxic agents, such as HgCl2, glycerol, cisplatin, gentamicin etc., have been known to exert cytotoxic effects on renal tubular cells [1, 2], inducing acute renal cell injury. However, as their cytotoxic action yet remains elusive, the preventive or therapeutic modalities for renal cell injury have not been established. Nevertheless, the accumulating data suggest that the involvement of oxidative stress (generation of oxygen free radicals) could be crucial for such renal cell injury induced by nephrotoxins [3, 4]. In addition, we have recently reported that oxidative stress would play a primary role in renal cell injury with certain nephrotoxins [5]. We were thus encouraged to further expand our study of nephrotoxin-mediated oxidative stress, focusing on biochemical parameters in the different renal cell line.
One of such biochemical parameters is glyoxalase I (Gly-I), a vital enzyme involved in the cellular detoxification process [6], playing a critical role in detoxifying and scavenging cytotoxic agents and metabolites (including free radicals) [7, 8]. Since activation of Gly-I inevitably requires reduced glutathione (GSH) as a cofactor [7], it is also categorized in a family of GSH-dependent enzymes. It is then plausible that Gly-I could be somewhat involved in detoxification of nephrotoxins.
A family of heat shock proteins (Hsps) is another interesting biochemical parameter, consisting of several species [9]. As they are functionally known as stress-response proteins, immediately responding to a variety of stresses exerted on the cells, they have been considered to play an important role in the cellular defense mechanism [10]. Among those Hsps, we are particularly interested in heat shock protein 90 (Hsp90) because it is primarily localized in the distal tubules and collecting ducts in the kidney [11] and would serve to protect renal cells from stress-related assaults [12]. It is thus possible that Hsp90 may also help protect renal cells from certain nephrotoxins.
We proposed that nephrotoxins would exert oxidative stress on renal cells as their primary cytotoxic action, triggering a cascade of biochemical events and leading ultimately to cell death. Accordingly, glycerol (GLC), a nephrotoxin capable of inducing renal cell injury, was chosen for studying its cytotoxic action on renal proximal tubular OK cells [13] in vitro (instead of LLC-PK1 cells used in our previous study). We first examined if GLC would actually exert oxidative stress on OK cells, and possible effects of GLC (through oxidative stress) were then examined on two biochemical parameters, Gly-I and Hsp90. We also explored if certain antioxidants (e.g. vitamins C/E and GSH) might provide cytoprotection against GLC-mediated oxidative assault. Detailed studies and notable findings are described and discussed herein.
| Materials and Methods | ▴Top |
Cell culture
The renal proximal tubular OK cells [13] were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). For experiments, OK cells were seeded at 2 × 105 cells/ml in 6-well plates or T-75 flasks and cultured with varying concentrations of glycerol (GLC). Cell number/viability was determined at specified times by the trypan blue exclusion method.
Lipid peroxidation (LPO) assay
Severity of oxidative stress on the cells was assessed by LPO assay measuring the amount of malondialdehyde (MDA) formed, which was indicative of oxidative damage in the plasma membrane [14]. The detailed procedures were described in the vendor’s protocol (Calbiochem, La Jolla, CA), and the amount of MDA formed was expressed by μM determined from the MDA standards.
Glyoxalase I (Gly-I) assay
Gly-I activity was measured following the method of Ranganathan and Tew [15]. After preparation of the reaction mixture (200 mM imidazole HCl, pH 7.0, 16 mM MgSO4, 7.9 mM methylglyoxal, 1 mM GSH), the reaction was started by the addition of cell lysates (40 μg). Due to a production of S-D-lactoylglutathione (E240 = 3.37 mM-1· cm-1), the increase in absorbance at 240 nm was measured with times on a spectrophotometer. Gly-I activity was then expressed by units/mg protein where one unit was defined to catalyze the formation of one μmol of S-D-lactoylglutathione per min.
Western blot analysis
The procedures essentially followed the protocol described previously [16]. Briefly, an equal amount of cell lysates (7 μg) obtained from control and agent-treated cells was subjected to 10% SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The blot (membrane) was incubated with the primary antibody against Hsp90 (anti-Hsp90), followed by an incubation with the secondary antibody conjugate. The immunoreactive protein bands were then detected by chemiluminescence following the manufacturer’s protocol (Kirkegaard and Perry Laboratories, Gaithersberg, MD).
Statistical analysis
All data were presented as mean ± SD (standard deviation), and statistical differences between groups were assessed with either one-way analysis of variance (ANOVA) or the unpaired Student’s t test. Values of P < 0.05 were considered to indicate statistical significance.
| Results | ▴Top |
Effects of glycerol (GLC) on OK cell proliferation
OK cells were cultured with varying concentrations (0 - 3%) of GLC and cell growth/viability was determined in 24 hours. GLC was capable of inducing 38%, 60%, 79%, 95%, and 100% growth reduction with 1%, 1.5%, 2%, 2.5%, and 3% GLC, respectively (Fig. 1). Although no apparent cell death was observed up to 2% GLC, a 95% growth reduction attained with 2.5% GLC was largely attributed to severe cell death. We then used this 2.5% GLC as the most effective concentration in the rest of our study.
 Click for large image | Figure 1. Dose-dependent effects of glycerol (GLC) on OK cell growth. Cells were cultured with varying concentrations (0, 0.5, 1, 1.5, 2, 2.5, or 3%) of GLC for 24 hours, and cell numbers were determined as mean ± SD (standard deviation) from three separate experiments (*P < 0.03). |
| Discussion | ▴Top |
As our previous study [5] suggested a primary role of oxidative stress in renal cell injury induced by nephrotoxins, we further investigated possible alterations in specific biochemical events induced by a nephrotoxin, glycerol (GLC), in renal tubular OK cells in vitro. GLC (2.5%) demonstrated to be highly cytotoxic, inducing severe cell death (95%) in 24 hours. Such cytotoxicity appeared to be primarily due to oxidative stress through GLC, indicated by LPO assay (Table 1). It is thus conceivable that such oxidative stress may trigger a cascade of various biochemical events, leading ultimately to cell death.
We next examined if certain antioxidant(s) might counteract with GLC-mediated oxidative stress. Interestingly, only NAC was found to protect OK cells from GLC oxidative assault (Fig. 2), implying the possible involvement of specific NAC-activated enzyme such as glyoxalase I (Gly-I) [6]. Our study then showed that a 75% loss in Gly-I activity after 6-hour GLC exposure (Fig. 3A) was completely reversed or prevented with NAC (Fig. 3B) and cell viability also remained at nearly 100% (Fig. 2). Thus, these results suggest that Gly-I may somewhat detoxify/diminish GLC cytotoxicity, contributing to NAC-provided cytoprotection.
It is crucial but remains uncertain how GLC would inactivate Gly-I. Yet, we assume that GLC-induced Gly-I inactivation may likely result from an insufficient availability of cellular GSH (synthesized in renal cells) because activation of Gly-I essentially requires GSH (or NAC used in this study) [7]. It is possible that GLC by itself or GLC-mediated oxidative stress may interfere with de novo synthesis of GSH, depriving cellular GSH. For example, γ-glutamylcysteine synthetase [18], a key enzyme involved in GSH synthesis, or other enzymes could be primarily targeted by GLC or free radicals. Further studies are thus required.
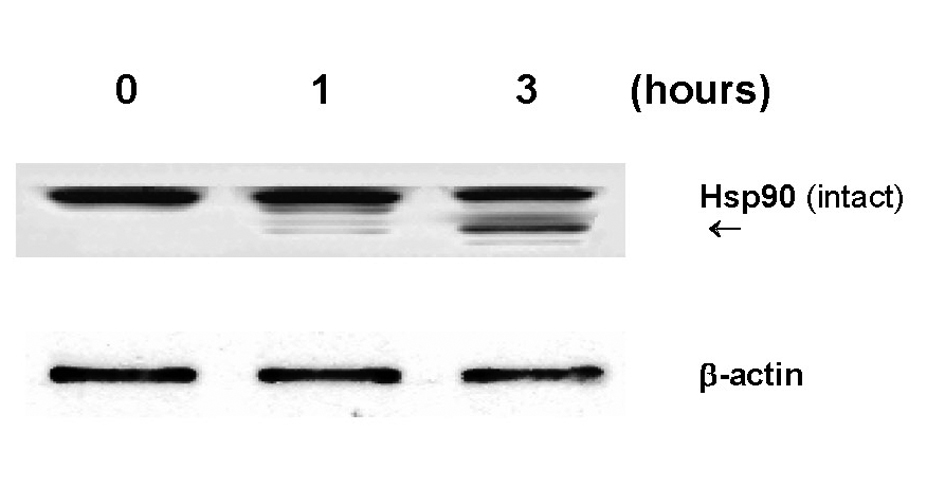
Another interesting biochemical alteration through GLC was partial degradation of Hsp90 (Fig. 4). As Hsp90 is shown to play a defensive role, its degradation could result in a weakening or destabilization of the cellular defense system, ultimately leading to renal cell death. It is yet of interest to further explore how Hsp90 would be degraded by GLC. As protein degradation is generally known to be carried out by protease(s), we assume that proteasome, a multicatalytic protease, might be involved in Hsp90 degradation because it has been shown to play a major role in degradation of intracellular proteins including Hsps [19]. Such study is currently underway in our laboratory.
In summary, the cytotoxic action of GLC primarily involves oxidative stress, causing adverse biochemical alterations such as inactivation of Gly-I and partial degradation of Hsp90. This indicates a collapse of the cellular detoxification and defense systems, presumably leading to renal cell death. However, NAC-provided cytoprotection against GLC, restoring Gly-I activity and protecting Hsp90 integrity, may have clinical implications in preventing renal cell injury/death induced by nephrotoxins.
| References | ▴Top |
- Natochin YV, Bakhteeva VT, Karpenko LA. Renal failure and nephrotoxic drug-induced disturbances in rat kidney tissue. Renal failure. 1994;16(6):687-696.
pubmed - Baliga R, Ueda N, Walker PD, Shah SV. Oxidant mechanisms in toxic acute renal failure. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1997;29(3):465-477.
pubmed - Diamond JR. The role of reactive oxygen species in animal models of glomerular disease. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1992;19(3):292-300.
pubmed - Andreoli SP, McAteer JA. Reactive oxygen molecule-mediated injury in endothelial and renal tubular epithelial cells in vitro. Kidney international. 1990;38(5):785-794.
pubmed - Louie B, Rajamahanty S, Pyo P, Choudhury M, Konno S. Mode of cytotoxic action of nephrotoxic agents: oxidative stress and glutathione-dependent enzyme. BJU international. 2010;105(2):264-268.
pubmed - Thornalley PJ. Glutathione-dependent detoxification of alpha-oxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chemico-biological interactions. 1998;111-112:137-151.
pubmed - Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer research. 1994;54(16):4313-4320.
pubmed - Volm M, Kastel M, Mattern J, Efferth T. Expression of resistance factors (P-glycoprotein, glutathione S-transferase-pi, and topoisomerase II) and their interrelationship to proto-oncogene products in renal cell carcinomas. Cancer. 1993;71(12):3981-3987.
pubmed - Ohtani H, Wakui H, Komatsuda A, Satoh K, Miura AB, Itoh H, Tashima Y. Induction and intracellular localization of 90-kilodalton heat-shock protein in rat kidneys with acute gentamicin nephropathy. Laboratory investigation; a journal of technical methods and pathology. 1995;72(2):161-165.
pubmed - Fukuda A, Osawa T, Oda H, Tanaka T, Toyokuni S, Uchida K. Oxidative stress response in iron-induced acute nephrotoxicity: enhanced expression of heat shock protein 90. Biochemical and biophysical research communications. 1996;219(1):76-81.
pubmed - Matsubara O, Kasuga T, Marumo F, Itoh H, Tashima Y. Localization of 90-kDa heat shock protein in the kidney. Kidney international. 1990;38(5):830-834.
pubmed - Hightower LE. Heat shock, stress proteins, chaperones, and proteotoxicity. Cell. 1991;66(2):191-197.
pubmed - Koyama H, Goodpasture C, Miller MM, Teplitz RL, Riggs AD. Establishment and characterization of a cell line from the American opossum (Didelphys virginiana). In vitro. 1978;14(3):239-246.
pubmed - Dargel R. Lipid peroxidation--a common pathogenetic mechanism. Experimental and toxicologic pathology: official journal of the Gesellschaft fur Toxikologische Pathologie. 1992;44(4):169-181.
pubmed - Ranganathan S, Tew KD. Analysis of glyoxalase-I from normal and tumor tissue from human colon. Biochimica et biophysica acta. 1993;1182(3):311-316.
pubmed - Mordente JA, Konno S, Chen Y, Wu JM, Tazaki H, Mallouh C. The effects of brefeldin A (BFA) on cell cycle progression involving the modulation of the retinoblastoma protein (pRB) in PC-3 prostate cancer cells. The Journal of urology. 1998;159(1):275-279.
pubmed - Wongmekiat O, Thamprasert K, Lumlertgul D. Renoprotective effect of trolox against ischaemia-reperfusion injury in rats. Clinical and experimental pharmacology & physiology. 2007;34(8):753-759.
pubmed - Issels RD, Nagele A, Eckert KG, Wilmanns W. Promotion of cystine uptake and its utilization for glutathione biosynthesis induced by cysteamine and N-acetylcysteine. Biochemical pharmacology. 1988;37(5):881-888.
pubmed - Grune T, Reinheckel T, Joshi M, Davies KJ. Proteolysis in cultured liver epithelial cells during oxidative stress. Role of the multicatalytic proteinase complex, proteasome. The Journal of biological chemistry. 1995;270(5):2344-2351.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.