

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Case Report
Volume 3, Number 2, April 2011, pages 85-92
Atypical Teratoid/Rhabdoid Tumors in Adults: A Case Report and Treatment-Focused Review
Nicole A. Shonkaa, g, Terri S. Armstrongb, Sujit S. Prabhuc, Amanda Childressd, Shauna Choie, Lauren A. Langfordf, Mark R. Gilbertb
aDivision of Oncology and Hematology, University of Nebraska Medical Center, 987680 Nebraska Medical Center, Omaha NE 68198-7680, USA
bDepartment of Neuro-Oncology, University of Texas at MD Anderson Cancer Center, 1400 Holcombe, Unit 431, Houston, Texas 77030, USA
cDepartment of Neurosurgery, University of Texas MD Anderson Cancer Center, 1400 Holcombe, Unit 442, Houston, Texas 77030, USA
dBrain and Spine Center, University of Texas at MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030, USA
eDivision of Pharmacy, Department of Neuro-Oncology, University of Texas at MD Anderson Cancer Center, 1400 Holcombe, Unit 431, Houston, Texas 77030, USA
fDepartment of Pathology/Neuropathology, University of Texas at MD Anderson Cancer Center, 1400 Holcombe, Houston, Texas 77030, USA
gCorresponding author: Nicole A. Shonka, Email:
Manuscript accepted for publication March 10, 2011
Short title: Atypical Teratoid/Rhabdoid Tumors in Adults
doi: https://doi.org/10.4021/jocmr535w
| Abstract | ▴Top |
Atypical teratoid/rhabdoid tumor is predominantly a childhood tumor and has only been rarely reported in adults; therefore, treatment regimens are often extrapolated from the pediatric experience. Typically, children are treated with craniospinal radiation therapy which is often followed by systemic chemotherapy. Employing pediatric regimens to treat this tumor in adult patients poses a particular risk for myelosuppression, as the prescribed doses in pediatric protocols exceed those tolerated by adults, and conventional craniospinal radiation can be associated with prolonged myelotoxicity and a depletion of the bone marrow reserve in vertebrae of adults. Here we present a case of a woman with a pineal region atypical teratoid/rhabdoid tumor, an unusual adult cancer presenting in an atypical location. This is followed by a review of the disease in adult patients with an emphasis on treatment and suggestions to minimize myelotoxicity.
Keywords: Atypical rhabdoid tumor; AT/RT; Pineal tumor; Adult
| Case Report | ▴Top |
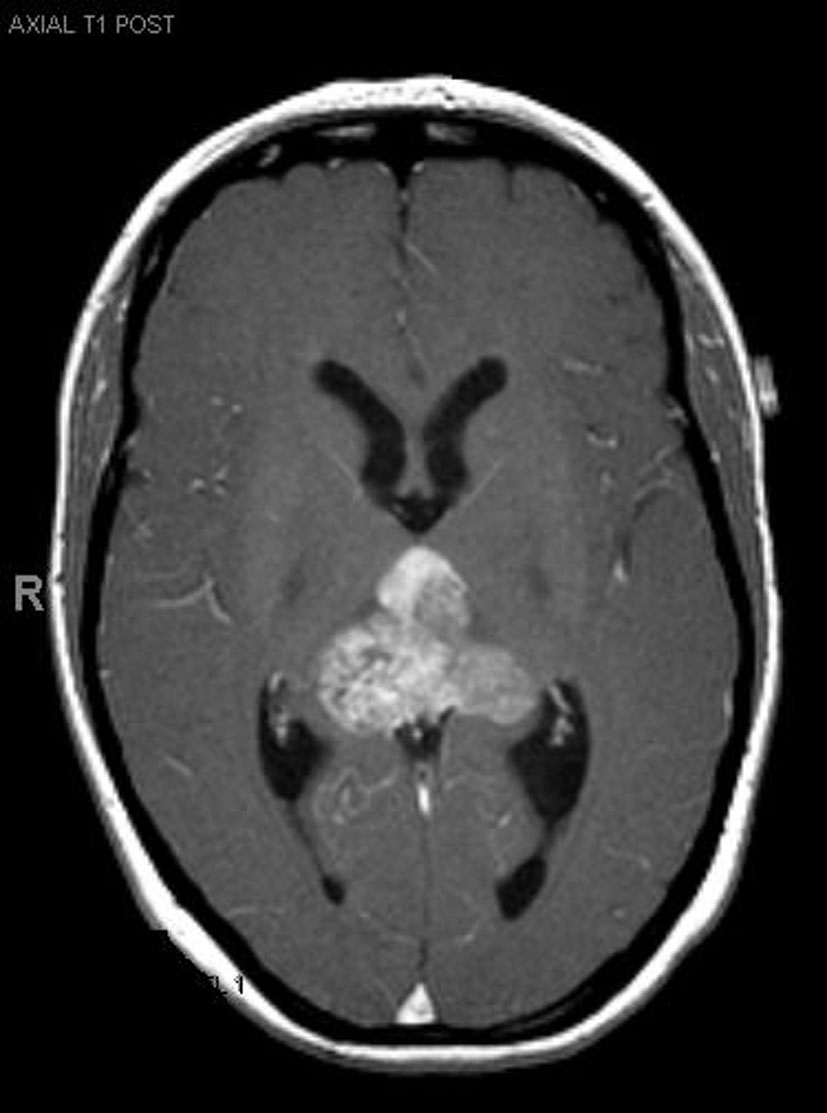 Click for large image | Figure 1.. Pre-operative MRI. |
A 33-year-old right-handed woman developed the sensation of fullness in her head followed months later by blurred vision that progressed to double vision. Brain MRI identified a large pineal mass and hydrocephalus (Fig. 1). A subtotal resection of the mass with concurrent placement of a ventriculoperitoneal shunt was performed at an outside institution (Fig. 2). Pathology suggested an epithelioid neoplasm, but a definitive diagnosis could not be made. Three weeks later, the tumor had regrown to its original size and a repeat supracerebellar infratentorial craniotomy was performed at MD Anderson Cancer Center with a near complete resection of the pineal mass (Fig. 3). Immunohistochemistry was positive for epithelial membrane antigen and smooth muscle actin. An antibody against the hSNF5/INI1 protein was negative in tumor cell nuclei. These findings confirmed the diagnosis of atypical teratoid/rhabdoid tumor (AT/RT), WHO grade IV. Cerebrospinal fluid and spinal MRIs were negative for tumor dissemination.
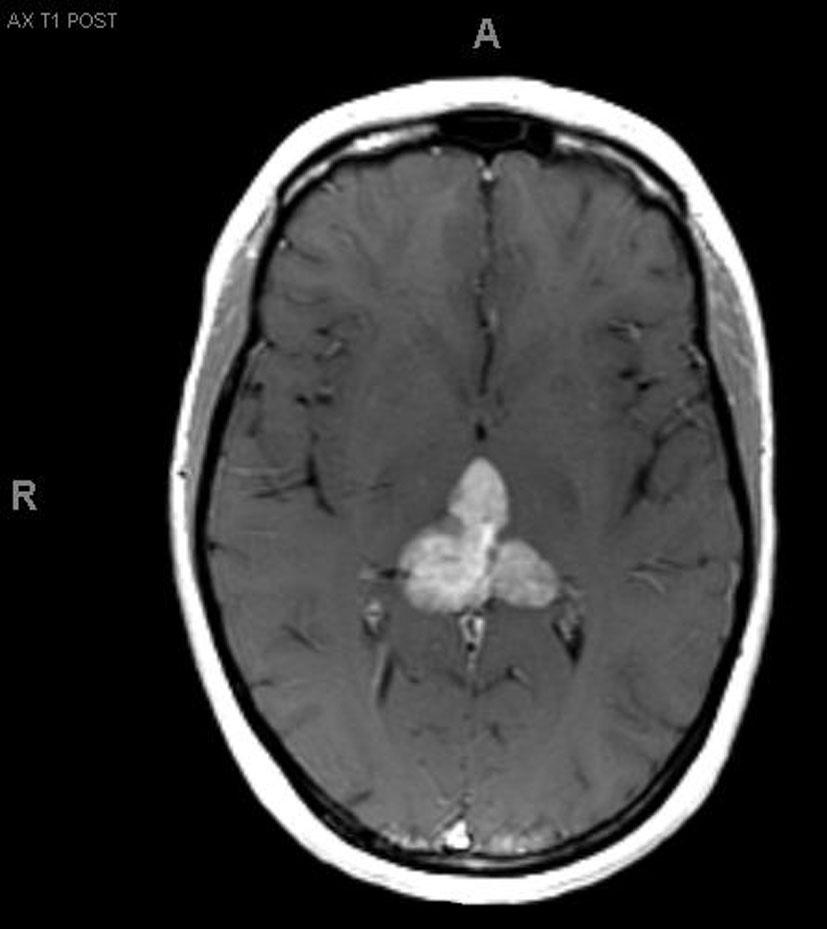 Click for large image | Figure 2.. MRI after first resection. |
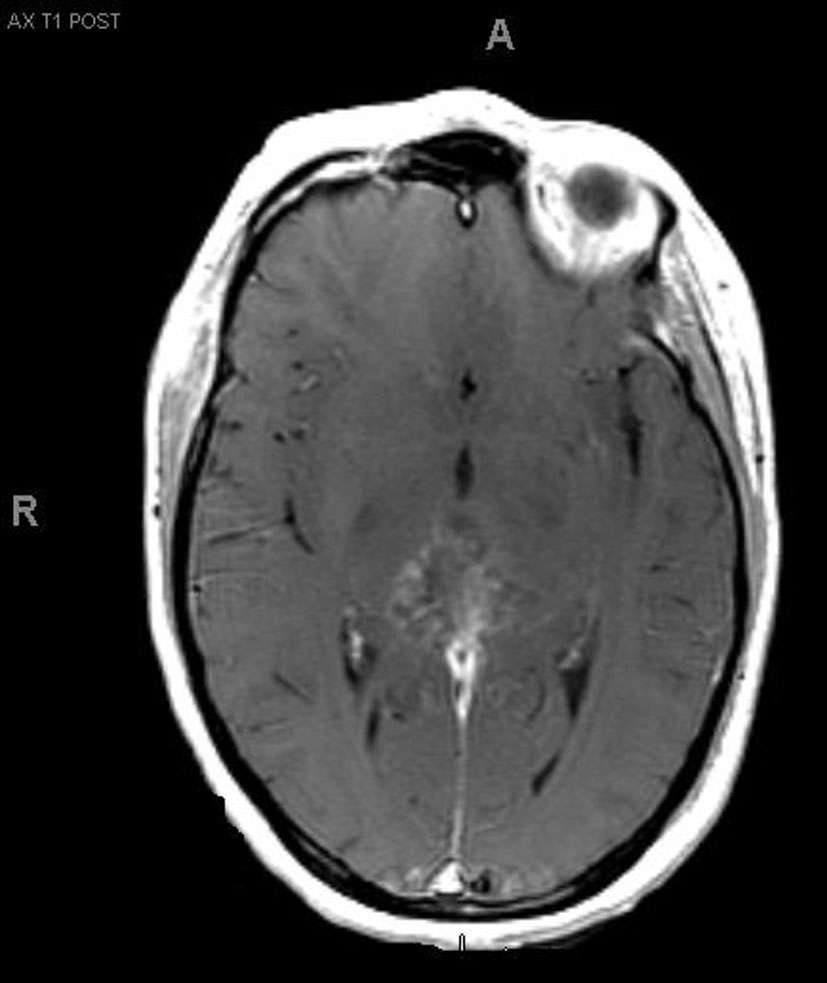 Click for large image | Figure 3.. MRI after second resection. |
Peripheral blood stem collection was performed prior to the initiation of chemotherapy. The patient underwent craniospinal radiation therapy (CSRT) and post-radiation MRI showed a modest decrease in the size of the residual tumor. Chemotherapy with Ifosphamide, Etoposide and Carboplatin (ICE) was given in 3-week cycles. After two cycles, brain MRI showed a partial tumor response. After her eighth cycle there was evidence of tumor progression with recurrence in the thalamus as well as along the occipital horn of the lateral ventricle. The treatment regimen was changed to doxorubicin, vincristine and temozolomide. The patient continues on this treatment regimen, and remains clinically and radiographically stable 18 months after the initial diagnosis.
| Review of the Literature | ▴Top |
The history of AT/RT
Beckwith and Palmer, in 1978, first coined the term ‘rhabdoid tumor’ to describe a histological variant of Wilm’s tumor found primarily in infants that was associated with an extremely poor prognosis [1]. The name was derived from its similarity in gross tumor appearance to a rhabdomyosarcoma; however, the cells differed from the expected morphological and immunohistochemical features of muscle [2]. A tumor composed of rhabdoid cells in the central nervous system (CNS), was first reported in 1985 [3]. The name ‘atypical teratoid/rhabdoid tumor’ (AT/RT) exemplifies the tumors disparate mixtures of rhabdoid, primitive neuroepithelial, mesenchymal and epithelial components [4]. AT/RT is much more frequently seen in infants and young children than older children and is rare in adults. AT/RT has an overall incidence of 1 - 2% of all brain tumors in children [4, 5]. They are estimated to account for over 10% of CNS tumors in infants, with a male preponderance up to the age of 3 which then seems to disappear [6, 7]. There exists a rhabdoid tumor predisposition syndrome which can be inherited in an autosomal dominant fashion, but most commonly occurs sporadically. The genetic form of AT/RT results from a germline loss of function mutations in INI1, also known as SMARCB1, a tumor suppressor gene at 22q11.23 [8]. This syndrome commonly manifests in tumors of the kidneys, brain and soft tissues. There have been just over 30 adult cases reported in the literature to date [9-31] (summarized in Table 1). Clinical presentation varies with tumor location in adults where a variety of primary locations have been reported.
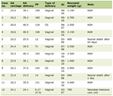 Click to view | Table 1. Adult Patients With AT/RT in the CNS |
No data exists to support imaging characteristics that differentiate AT/RTs from other primitive neuroectodermal tumors [32]. Report from the AT/RT workshop in 2002 noted that half of all AT/RTs are in the posterior fossa, although the tumor has been noted throughout the nervous system and in extramedullary sites. Tumors can be extraaxial and invade adjacent structures such as the meninges as well [32]. In adults, these are primarily found in the cerebral hemispheres [33] and are rare in the cerebellum and spinal cord [16, 17, 19, 21, 25]. Similar to our case, two other adult AT/RTs have been found in the pineal region [23, 34].
Computed tomography (CT) usually shows a hyperdense mass that intensely enhances after administration of intravenous contrast. On T1-weighted magnetic resonance imaging (MRI) the mass is commonly isointense with hyperintense areas that result from intratumoral bleeding. The T2 imaging is more heterogenous with hypointense to hyperintense areas indicating a mixture of necrosis, hemorrhage, cystic changes, and calcifications [32, 35]. Peritumoral edema was variable in the meta-analysis of 133 patients done by Oka [36]. MR spectroscopy shows a marked elevation of choline and low or absent N-acetylaspartate (NAA) and creatinine, as would be expected. In a review of thirteen patients ages 4 months to 15 years with AT/RT, all tumors except one enhanced with contrast on MRI [35].
AT/RTs consist of a combination of rhabdoid, primitive neuroepithelial, mesencymal and epithelial cells. Approximately one-third contain epithelial or mesenchymal cells, and only 10% are comprised purely of rhabdoid cells [32]. This heterogeneity makes the discrimination between AT/RT and the other tumors of embryonal tissue, namely medulloblastoma and primitive neuroectodermal tumor (PNET), difficult using histologic criteria [37, 38]. Thus far, the histogenesis of this tumor has remained elusive [10, 32, 39]. Proliferative activity is high, and Ki-67/MIB-1 labelling indices averaged 63.9% in a series of pediatric patients [40], and have ranged from less than 20% to 80% [9, 12, 16, 34].
Immunohistochemistry (IHC) is positive for rhabdoid cell markers including epithelial membrane antigen (EMA), vimentin, and smooth-muscle actin (SMA) in the majority of tumors and markers for germ-cell tumors such as alpha-fetoprotein and placental alkaline phosphatase are consistently negative [4, 32]. The tumors may also express glial fibrillary acidic protein, keratin, synaptophysin, and neurofilament protein [32]. IHC staining for the INI1 protein, a component of a SWI/SNF ATP-dependent chromatin-remodeling complex has been shown to be highly sensitive and specific for AT/RTs [41-43]. Versteege suggested that any loss-of-function mutations of INI1 contribute to oncogenesis after noting bi-allelic alterations of INI1 (i.e., any truncating mutation of one allele caused loss of the other allele). Monosomy 22 or deletions of chromosome band 22q11 are found in most AT/RTs, however, alterations of chromosome 22 are shared in other CNS tumors as well. PNETs may have deletions of chromosome 22 but can sometimes be differentiated from AT/RTs by the presence of chromosome 17 abnormalities [4, 42, 44].
Prognosis
Tekautz and colleagues reported the outcomes from a series of 37 pediatric patients [7]. Event-free survival (EFS) and overall survival (OS) at two years for children aged three years or older was 78% and 89% vs 11% and 17 % for younger children. Oka performed a meta-analysis of 133 patients and found that 98 (74%) patients had succumbed to their disease within 24 months of diagnosis, with a mean OS of 8.5 months. Seventy-five percent of these patients were younger than three years of age [36]. Recently, the modified IRS-III regimen evaluated by Chi and colleagues increased two-year survival to 70% ± 10% compared to historic median survival of only 6 - 11 months [32, 39, 45]. Of the 31 adult patients whose survival data were reported in the literature, the median survival was 15 - 18 months, although it ranged widely from two weeks to over 17 years (Table 1).
Treatment
The impact of the extent of surgical resection on outcome has not been fully studied. Packer reported from the pediatric AT/RT registry an OS of 8.5 months that lengthened to 13 months in patients who had gross total resection (GTR) of their tumors. In this report, a personal communication with J. Hilden M.D. is cited that of the eight patients in the registry with an OS of greater than 18 months, six of those had undergone a GTR [32]. Hilden’s report favors more aggressive resection as well with an OS of 20 months vs 15.25 months [46].
Click to view | Table 2. Chemotherapy Used for AT/RT in Children |
Treatment paradigms for adult patients have been extracted from the pediatric literature. Chemotherapeutic regimens used in the pediatric population vary, but regimens commonly utilize vincristine with an alkylating and a platinum agent. Table 2 highlights several regimens and outcomes in pediatric patients.
Excluding our patient, there have been 31 adult cases reported in the (English) literature. There is no information available regarding treatment given to three of these patients, and no survival information was provided for three patients. Of the 28 adult patients in whom treatment was reported, 14 (50%) received chemotherapy, either concurrent with or after radiation therapy. Temozolomide and ICE were commonly used. Survival in patients who received chemotherapy ranged from 6 months to 17 years, with a median survival of 24 months. Those who received surgery and radiation therapy without chemotherapy had a survival between 2 and 7 years, with a median survival of 9 months. In a small case series, we cannot confirm the superiority of one regimen or even a benefit from chemotherapy. The data supports the importance of preserving bone marrow function so that systemic chemotherapy remains a viable option.
Much of the data regarding treatment has focused on chemotherapy, since the majority of patients diagnosed with AT/RT are under two to three years old when RT is avoided if possible. Those patients over the age of three are routinely given RT, often in the form of CSRT as leptomeningeal disease (LMD) is often present at diagnosis and is common at recurrence. The AT/RT registry shows a high rate of local recurrence, and those who survived more than 18 months were more likely (75%) to have received RT [32]. Patients are given varying doses between 40 - 60 Gy, and stereotactic radiosurgery has been used for recurrent disease when resection is not feasible [47]. There are no data on the response to RT in adult AT/RT but of 13 patients in a case series from the Childrens Hospital of Philadelphia, only two patients had an objective response [4].
In the report on 42 pediatric cases by Hilden, 13 patients underwent stem cell rescue as part of their primary treatment, which underscores the significant myelosuppression of treatment regimens for AT/RT [46]. Approximately 40% of adult bone marrow resides in the spine [48]. In our patient, we were concerned about the additive myelotoxicity of CSRT and chemotherapy and so a stem cell harvest was performed and the patient was given radiation with protons. Unlike conventional photon radiotherapy, proton radiotherapy focuses the maximum dose to the target tissue while sparing normal tissues from much of the entry dose and the entire exit dose. This occurs as protons lose only a small amount of their energy in tissue until they reach the target tissue, after which the residual energy is rapidly lost, resulting in a steep treatment gradient [49]. Therefore, the use of proton-beam radiation for craniospinal treatment may allow a partial sparing of vertebral body radiation exposure, lessening the impact on this major component of hematopoeisis and lessening the degree of myelotoxcity.
Laboratory studies have focused on establishing an AT/RT in vitro cell culture model on which preclinical studies can investigate both chemotherapeutic agents as well as targeted therapies [50-53]. Insulin-growth factor-1 receptor (IGF-1R) inhibition has been shown to sensitize cells to both chemotherapy and radiation [53]. Using AT/RT cells cultured from CSF, Narendran noted growth inhibition with low concentrations of arsenic trioxide, Prima-1 (targets mutant p53 proteins), oxaliplatin, cisplatin and rebeccamycin. Thalidomide, etoposide, cytarabine and paclitaxel had intermediate MICs. The optimal treatment remains to be defined, but the increasing recognition of this disease and the development of good laboratory models will hopefully accelerate therapeutic advances.
Conclusion
AT/RT remains a rare adult disease. However, as our knowledge of AT/RTs increases we anticipate that there will be more standardization of treatment. Aggressive resection followed by multimodality treatment appears to yield more long-term survivors. In adults, although the use of RT does not convey the same devastating developmental arrest, there are still reasons to minimize the effects to normal tissues particularly bone marrow, supporting the use of proton radiation, particularly since CSRT is a standard treatment. Although the optimal chemotherapy regimen has not been defined for adults with AT/RT, several regimens have been used with evidence of activity. Further advances in treatment will likely require more laboratory studies generating novel treatment regimens for clinical trial testing. The rarity of adult AT/RT suggests that treatment regimens will continue to rely on advances in pediatric treatments.
| Conflicts of Interest | ▴Top |
The authors have no relevant conflicts of interest to disclose.
| References | ▴Top |
- Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer. 1978;41(5):1937-1948.
pubmed - Haas JE, Palmer NF, Weinberg AG, Beckwith JB. Ultrastructure of malignant rhabdoid tumor of the kidney. A distinctive renal tumor of children. Hum Pathol. 1981;12(7):646-657.
pubmed - Montgomery P, Kuhn JP, Berger PE. Rhabdoid tumor of the kidney: a case report. Urol Radiol. 1985;7(1):42-44.
pubmed - Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85(1):56-65.
pubmed - Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001;17(9):503-511.
pubmed - Allen JC, Judkins AR, Rosenblum MK, Biegel JA. Atypical teratoid/rhabdoid tumor evolving from an optic pathway ganglioglioma: case study. Neuro Oncol. 2006;8(1):79-82.
pubmed - Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, Krasin M,
et al . Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol. 2005;23(7):1491-1499.
pubmed - Swensen JJ, Keyser J, Coffin CM, Biegel JA, Viskochil DH, Williams MS. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet. 2009;46(1):68-72.
pubmed - Horn M, Schlote W, Lerch KD, Steudel WI, Harms D, Thomas E. Malignant rhabdoid tumor: primary intracranial manifestation in an adult. Acta Neuropathol. 1992;83(4):445-448.
pubmed - Fisher BJ, Siddiqui J, Macdonald D, Cairney AE, Ramsey D, Munoz D, Del Maestro R. Malignant rhabdoid tumor of brain: an aggressive clinical entity. Can J Neurol Sci. 1996;23(4):257-263.
pubmed - Ashraf R, Bentley RC, Awan AN, McLendon RE, Ragozzino MW. Implantation metastasis of primary malignant rhabdoid tumor of the brain in an adult (one case report). Med Pediatr Oncol. 1997;28(3):223-227.
pubmed - Byram D. Regarding Weiss et al., IJROBP 41:103-109; 1998. Int J Radiat Oncol Biol Phys. 1999;45(1):247.
pubmed - Kuge A, Kayama T, Tsuchiya D, Kawakami K, Saito S, Nakazato Y, Suzuki H. [Suprasellar primary malignant rhabdoid tumor in an adult: a case report]. No Shinkei Geka. 2000;28(4):351-358.
pubmed - Arrazola J, Pedrosa I, Mendez R, Saldana C, Scheithauer BW, Martinez A. Primary malignant rhabdoid tumour of the brain in an adult. Neuroradiology. 2000;42(5):363-367.
pubmed - Lutterbach J, Liegibel J, Koch D, Madlinger A, Frommhold H, Pagenstecher A. Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol. 2001;52(1):49-56.
pubmed - Bruch LA, Hill DA, Cai DX, Levy BK, Dehner LP, Perry A. A role for fluorescence in situ hybridization detection of chromosome 22q dosage in distinguishing atypical teratoid/rhabdoid tumors from medulloblastoma/central primitive neuroectodermal tumors. Hum Pathol. 2001;32(2):156-162.
pubmed - Pimentel J, Silva R, Pimentel T. Primary malignant rhabdoid tumors of the central nervous system: considerations about two cases of adulthood presentation. J Neurooncol. 2003;61(2):121-126.
pubmed - Kawaguchi T, Kumabe T, Watanabe M, Tominaga T. Atypical teratoid/rhabdoid tumour with leptomeningeal dissemination in an adult. Acta Neurochir (Wien). 2004;146(9):1033-1038, discussion 1038.
pubmed - Kachhara R, Retnam TM, Kumar S, Nair S, Bhattacharya RN, Krishnamoorthy T, Radhakrishnan VV. Rhabdoid tumor of the thalamus. Neurol India. 2003;51(2):273-274.
pubmed - Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A. Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol. 2005;15(1):23-28.
pubmed - Erickson ML, Johnson R, Bannykh SI, de Lotbiniere A, Kim JH. Malignant rhabdoid tumor in a pregnant adult female: literature review of central nervous system rhabdoid tumors. J Neurooncol. 2005;74(3):311-319.
pubmed - Ingold B, Moschopulos M, Hutter G, Seeger H, Rothlisberger B, Landolt H, Yonekawa Y,
et al . Abdominal seeding of an atypical teratoid/rhabdoid tumor of the pineal gland along a ventriculoperitoneal shunt catheter. Acta Neuropathol. 2006;111(1):56-59.
pubmed - Rezanko T, Tunakan M, Kahraman A, Sucu HK, Gelal F, Akkol I. Primary rhabdoid tumor of the brain in an adult. Neuropathology. 2006;26(1):57-61.
pubmed - Zarovnaya EL, Pallatroni HF, Hug EB, Ball PA, Cromwell LD, Pipas JM, Fadul CE,
et al . Atypical teratoid/rhabdoid tumor of the spine in an adult: case report and review of the literature. J Neurooncol. 2007;84(1):49-55.
pubmed - Samaras V, Stamatelli A, Samaras E, Stergiou I, Konstantopoulou P, Varsos V, Judkins AR,
et al . Atypical teratoid/rhabdoid tumor of the central nervous system in an 18-year-old patient. Clin Neuropathol. 2009;28(1):1-10.
pubmed - Cossu A, Massarelli G, Manetto V, Viale G, Tanda F, Bosincu L, Iuzzolino P,
et al . Rhabdoid tumours of the central nervous system. Report of three cases with immunocytochemical and ultrastructural findings. Virchows Arch A Pathol Anat Histopathol. 1993;422(1):81-85.
pubmed - Balaton AJ, Vaury P, Videgrain M. Paravertebral malignant rhabdoid tumor in an adult. A case report with immunocytochemical study. Pathol Res Pract. 1987;182(5):713-718.
pubmed - Chen YW, Wong TT, Ho DM, Huang PI, Chang KP, Shiau CY, Yen SH. Impact of radiotherapy for pediatric CNS atypical teratoid/rhabdoid tumor (single institute experience). Int J Radiat Oncol Biol Phys. 2006;64(4):1038-1043.
pubmed - Chacko G, Chacko AG, Dunham CP, Judkins AR, Biegel JA, Perry A. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neurooncol. 2007;84(2):217-222.
pubmed - Makuria AT, Rushing EJ, McGrail KM, Hartmann DP, Azumi N, Ozdemirli M. Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neurooncol. 2008;88(3):321-330.
pubmed - Arita K, Sugiyama K, Sano T, Oka H. Atypical teratoid/rhabdoid tumour in sella turcica in an adult. Acta Neurochir (Wien). 2008;150(5):491-495, discussion 496.
pubmed - Packer RJ, Biegel JA, Blaney S, Finlay J, Geyer JR, Heideman R, Hilden J,
et al . Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol. 2002;24(5):337-342.
pubmed - Judkins AR. Immunohistochemistry of INI1 expression: a new tool for old challenges in CNS and soft tissue pathology. Adv Anat Pathol. 2007;14(5):335-339.
pubmed - Sugita Y, Takahashi Y, Hayashi I, Morimatsu M, Okamoto K, Shigemori M. Pineal malignant rhabdoid tumor with chondroid formation in an adult. Pathol Int. 1999;49(12):1114-1118.
pubmed - Meyers SP, Khademian ZP, Biegel JA, Chuang SH, Korones DN, Zimmerman RA. Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. AJNR Am J Neuroradiol. 2006;27(5):962-971.
pubmed - Oka H, Scheithauer BW. Clinicopathological characteristics of atypical teratoid/rhabdoid tumor. Neurol Med Chir (Tokyo). 1999;39(7):510-517, discussion 517-518.
pubmed - Biegel JA, Fogelgren B, Zhou JY, James CD, Janss AJ, Allen JC, Zagzag D,
et al . Mutations of the INI1 rhabdoid tumor suppressor gene in medulloblastomas and primitive neuroectodermal tumors of the central nervous system. Clin Cancer Res. 2000;6(7):2759-2763.
pubmed - Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, Weissman B,
et al . The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res. 2002;62(1):323-328.
pubmed - Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, Duffner PK,
et al . Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998;22(9):1083-1092.
pubmed - Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H. Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol. 2000;99(5):482-488.
pubmed - Kalpana GV, Marmon S, Wang W, Crabtree GR, Goff SP. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science. 1994;266(5193):2002-2006.
pubmed - Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A,
et al . Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203-206.
pubmed - Biegel JA. Cytogenetics and molecular genetics of childhood brain tumors. Neuro Oncol. 1999;1(2):139-151.
pubmed - Biegel JA, Rorke LB, Packer RJ, Emanuel BS. Monosomy 22 in rhabdoid or atypical tumors of the brain. J Neurosurg. 1990;73(5):710-714.
pubmed - Chi SN, Zimmerman MA, Yao X, Cohen KJ, Burger P, Biegel JA, Rorke-Adams LB,
et al . Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol. 2009;27(3):385-389.
pubmed - Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, Walter AW,
et al . Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004;22(14):2877-2884.
pubmed - Olson TA, Bayar E, Kosnik E, Hamoudi AB, Klopfenstein KJ, Pieters RS, Ruymann FB. Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol. 1995;17(1):71-75.
pubmed - Russell WJ, Yoshinagah Antoku S, Mizuno M. Active bone marrow distribution in the adult. Br J Radiol. 1966;39(466):735-739.
pubmed - Yock TI, Tarbell NJ. Technology insight: Proton beam radiotherapy for treatment in pediatric brain tumors. Nat Clin Pract Oncol. 2004;1(2):97-103, quiz 101 p following 111.
pubmed - Furchert SE, Lanvers-Kaminsky C, Juurgens H, Jung M, Loidl A, Fruhwald MC. Inhibitors of histone deacetylases as potential therapeutic tools for high-risk embryonal tumors of the nervous system of childhood. Int J Cancer. 2007;120(8):1787-1794.
pubmed - Maris JM, Courtright J, Houghton PJ, Morton CL, Kolb EA, Lock R, Tajbakhsh M,
et al . Initial testing (stage 1) of sunitinib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;51(1):42-48.
pubmed - Narendran A, Coppes L, Jayanthan A, Coppes M, Teja B, Bernoux D, George D,
et al . Establishment of atypical-teratoid/rhabdoid tumor (AT/RT) cell cultures from disseminated CSF cells: a model to elucidate biology and potential targeted therapeutics. J Neurooncol. 2008;90(2):171-180.
pubmed - D'Cunja J, Shalaby T, Rivera P, von Buren A, Patti R, Heppner FL, Arcaro A,
et al . Antisense treatment of IGF-IR induces apoptosis and enhances chemosensitivity in central nervous system atypical teratoid/rhabdoid tumours cells. Eur J Cancer. 2007;43(10):1581-1589.
pubmed - Agranovich AL, Ang LC, Griebel RW, Kobrinsky NL, Lowry N, Tchang SP. Malignant rhabdoid tumor of the central nervous system with subarachnoid dissemination. Surg Neurol. 1992;37(5):410-414.
pubmed - Weinblatt M, Kochen J. Rhabdoid tumor of the central nervous system. Med Pediatr Oncol. 1992;20(3):258.
pubmed - Hanna SL, Langston JW, Parham DM, Douglass EC. Primary malignant rhabdoid tumor of the brain: clinical, imaging, and pathologic findings. AJNR Am J Neuroradiol. 1993;14(1):107-115.
pubmed - Dang T, Vassilyadi M, Michaud J, Jimenez C, Ventureyra EC. Atypical teratoid/rhabdoid tumors. Childs Nerv Syst. 2003;19(4):244-248.
pubmed - Izycka-Swieszewska E, Debiec-Rychter M, Wasag B, Wozniak A, Gasecki D, Plata-Nazar K, Bartkowiak J,
et al . A unique occurrence of a cerebral atypical teratoid/rhabdoid tumor in an infant and a spinal canal primitive neuroectodermal tumor in her father. J Neurooncol. 2003;61(3):219-225.
pubmed - Wharton SB, Wardle C, Ironside JW, Wallace WH, Royds JA, Hammond DW. Comparative genomic hybridization and pathological findings in atypical teratoid/rhabdoid tumour of the central nervous system. Neuropathol Appl Neurobiol. 2003;29(3):254-261.
pubmed - Zimmerman MA, Goumnerova LC, Proctor M, Scott RM, Marcus K, Pomeroy SL, Turner CD,
et al . Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor. J Neurooncol. 2005;72(1):77-84.
pubmed - Fidani P, De Ioris MA, Serra A, De Sio L, Ilari I, Cozza R, Boldrini R,
et al . A multimodal strategy based on surgery, radiotherapy, ICE regimen and high dose chemotherapy in atypical teratoid/rhabdoid tumours: a single institution experience. J Neurooncol. 2009;92(2):177-183.
pubmed - Gardner SL, Asgharzadeh S, Green A, Horn B, McCowage G, Finlay J. Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer. 2008;51(2):235-240.
pubmed - Lassaletta A, Lopez-Ibor B, Mateos E, Gonzalez-Vicent M, Perez-Martinez A, Sevilla J, Diaz MA,
et al . Intrathecal liposomal cytarabine in children under 4 years with malignant brain tumors. J Neurooncol. 2009;
pubmed - Biswas A, Goyal S, Puri T, Das P, Sarkar C, Julka PK, Bakhshi S,
et al . Atypical teratoid rhabdoid tumor of the brain: case series and review of literature. Childs Nerv Syst. 2009;
pubmed - Ertan Y, Sezak M, Turhan T, Kantar M, Ersahin Y, Mutluer S, Vergin C,
et al . Atypical teratoid/rhabdoid tumor of the central nervous system: clinicopathologic and immunohistochemical features of four cases. Childs Nerv Syst. 2009;25(6):707-711.
pubmed - Wang CH, Hsu TR, Wong TT, Chang KP. Efficacy of temozolomide for recurrent embryonal brain tumors in children. Childs Nerv Syst. 2009;25(5):535-541.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.