

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Original Article
Volume 11, Number 6, June 2019, pages 391-400
Humoral and Cellular Patterns of Early Endothelial Progenitor Cells in Relation to the Cardiovascular Risk in Axial Spondylarthritis
Susann Patschana, b, Maria Vogta, Donia Bakhtiaria, Carsten Peter Bramlagea, Elvira Henzea, Gerhard Anton Mullera, Andreas Krausec, Daniel Patschanb, d
aClinic of Nephrology and Rheumatology, University Hospital of Gottingen, Gottingen, Germany
bDepartment of Cardiology, Pulmonology, Angiology and Nephrology, Brandenburg Medical School, University Hospital Brandenburg, Brandenburg, Germany
cRheumatology and Clinical Immunology, Immanuel-Krankenhaus Berlin, Berlin, Germany
dCorresponding Author: Daniel Patschan, Department of Cardiology, Pulmonology, Angiology and Nephrology, Brandenburg Medical School, Hochstraße 29, 14770 Brandenburg an der Havel, Germany
Manuscript submitted April 10, 2018, accepted April 30, 2018
Short title: EPCs in SpA
doi: https://doi.org/10.14740/jocmr3441w
| Abstract | ▴Top |
Background: Spondylarthritis (SpA) significantly affects sacroiliac, intervertebral and peripheral joints. Patients with SpA suffer from increased cardiovascular risk (CVR). The endothelial progenitor cell (EPC) system critically perpetuates vascular repair. The aim of the study was to evaluate circulating EPCs in axial (ax)SpA with special attention on parameters of disease activity and CVR.
Methods: Disease activity and functional impairment were quantified in 50 axSpA patients by using standardized parameters (Bath ankylosing spondylitis disease activity index (BASDAI), C-reactive protein (CRP), finger-floor distance (FFD) and Ott’ sign). Circulating EPCs and EPC regeneration were analyzed (fluorescence-activated cell sorting (FACS) and colony-forming unit (CFU) assay). Serum vasomodulatory mediators were quantified by enzyme-linked immunosorbent assay (ELISA).
Results: EPC colony numbers were lower in axSpA as compared to controls. Females displayed more colonies than males. In addition, fewer colonies were observed in smokers, in patients with a BASDAI of below 4 and in hypertension. Circulating CD133+/KDR+ cells did not differ between the groups. Follow-up analysis (33 months later) did not show any differences in gender, colony formation, CD133+/KDR+ cells or serum levels of vasomodulatory mediators if related to the categories of BASDAI, Ott’ sign or FFD.
Conclusions: EPC colony formation is significantly affected in axSpA with particularly low levels in males. EPC-related parameters do not allow predicting disease activity-related or functional parameters nor are they useful for CVR assessment in SpA.
Keywords: SpA; EPCs; Cardiovascular risk
| Introduction | ▴Top |
Spondylarthritides (SpA) including ankylosing spondylitis (AS) and psoriatic arthritis (PsA) are characterized by a significantly higher than average cardiovascular risk (CVR) [1]. Recent data from the DESIR cohort showed increased prevalences of arterial hypertension in particular and of CVR in general in early axial spondylarthritis (axSpA) [2]. An investigation published in 2013 revealed higher carotid intima-media thickness (IMT) in 36 SpA patients as compared to healthy controls. In addition, IMT was associated with inflammatory markers, the Bath ankylosing spondylitis disease activity index (BASDAI) and certain clinical parameters such as axial or peripheral spine/joint affection, dactylitis and others [3]. Poddubnyy and Rebrov documented a positive correlation between AS disease activity and the risk for aquiring cardiovascular pathology as well [4]. The mentioned study also indicated endothelial dysfunction to significantly occur in AS individuals with high as opposed to those with low to moderate disease activity (endothelium-dependent vasodilation (EDVD)). A newer investigation, published in 2016, identified a positive correlation between the CVR and radiographic progression in axSpA [5]. Different established CVR factors (low-density lipoprotein (LDL), systolic blood pressure and Framingham risk score (FRS)) significantly differed between AS and controls. The grade of sacroiliitis and the numbers of syndesmophytes correlated with the FRS. As a matter of fact, the latter of these two parameters and the FRS were associated in an independent manner (multivariate analysis). Finally, Pederson and colleagues intensively investigated the impact of anti-tumor necrosis factor (TNF) treatment in axSpA, including numerous variables such as radiographic progression/inflammation and circulating parameters of inflammation, angiogenesis and cartilage/bone turnover [6]. Together, the mentioned studies suggest a dynamic relationship between SpA-associated inflammation/disease progression, atherogenesis and endothelial dysfunction.
For many years, neovascularization has been suggested to exclusively occur by the expansion of mature vessel wall cells such as smooth muscle and endothelial cells. This process, termed angiogenesis can also be supported by immature vascular wall precursor or progenitor cells. Endothelial progenitor cells (EPCs) have extensively been studied since the hallmark investigation published by Asahara in the late 1990s [7]. A lot of effort has been made in order to further characterize EPCs. Currently, two major EPC populations are being distinguished, early and late EPCs (eEPCs/lEPCs) [8-11]. Most of the research performed in recent years focused on eEPCs, showing that the cells are capable to prevent vertebrate organisms from ischemia-induced organ damage if administered in a systemic fashion [12-14]. Thus, the cells, although not acting in a direct manner [15, 16], presumably play a key role in endogenous vascular repair. In addition, the cells have been established as surrogate markers in ischemic diseases. In a relevant number of vascular diseases, either characterized by micro- or macrovascular dysfunction, eEPCs exhibit alterations in their proliferative capacity. Studies have been performed in coronary artery disease, chronic kidney dysfunction, sepsis, vasculitis and rheumatoid arthritis, respectively [17-20]. The data on EPCs in SpA are rare to put it mildly. One study showed peripheral EPC depletion in AS with lower cell numbers in negative correlation with the Bath ankylosing spondylitis disease activity index (BASDAI) and serum C-reactive protein (CRP) [21]. It needs to be noted that the authors presumably did not analyze cells that may be defined as EPCs in a broader sense, since they employed the two proteins CD34 and CD133 for cell detection. In reality, they most likely enumerated hematopoietic precursors. Thus, the data become even more limited.
Our study aimed to investigate peripheral circulating eEPCs, eEPC regeneration and certain humoral indicators of impaired angiogenesis in axSpA. The special interest focused on parameters of disease activity and atherogenic risk factors. In the article, we will from now on use the term EPCs instead of eEPCs.
| Materials and Methods | ▴Top |
Patients and setting
The presented investigation was a single-center analysis, performed at the Department of Nephrology and Rheumatology (University Hospital of Gottingen, Germany) initially between 2012 and 2014 with a follow-up visit 2 years later. The study was formally approved by the local ethics committee (name of the ethics committee: Medical Ethics Committee of the University Hospital of Gottingen - grant number: 17/02/08). Patients suffering from axSpA according to the ASAS criteria from 2009 [22] were screened and included in the study after obtaining written consent. Individuals with psoriatic arthritis, reactive arthritis and enteropathic arthritis were excluded from the study. Diagnoses of the former diseases were made by history and clinical examination. However, for further clinical characterization, especially for evaluating disease-associated decreases in mobility, several indices were employed: the test of Schober and Ott, finger-floor distance (FFD) and two well-established scores (Bath ankylosing spondylitis metrology index (BASMI) and Bath ankylosing spondylitis functional index (BASFI)). Disease activity was quantified by using the BASDAI and the ankylosing spondylitis disease activity score (ASDAS). The following cut-off values were used for defining activity: BASDAI < 4 indicates efficient disease control; 4 indicates insufficient disease control; ASDAS < 1.3 indicates mild activity; 1.3 - 2.1 indicates moderate activity; 2.1 - 3.5 indicates high activity; > 3.5 indicates very high activity. Laboratory parameters as well as human leukocyte antigen B27 (HLA-B27) analyses were collected/performed at the/from the database of the Clinic of Nephrology and Rheumatology of the University Hospital of Gottingen.
EPC quantification and regeneration
Quantification of peripheral circulating EPCs and analysis of EPC regeneration were performed as described extensively in previously published investigations [19, 20, 23]. However, the methodological approach shall be described in detail.
Circulating EPCs
Mononuclear cells (MNCs) were isolated by density gradient centrifugation using Histopaque-1077 solution (Sigma Diagnostics, St. Louis, MO, USA) from about 7.5 mL of heparinized peripheral blood. Cells were primarily incubated for 1 h on ice with one or more of the following antibodies: rabbit anti CD133 (ab16518; Abcam, Cambridge, UK), mouse anti-human VEGFR2 (KDR, directly conjugated - FAB 3571F; R&D Systems, Minneapolis, MN, USA), followed by secondary incubation with PE-conjugated goat anti-rabbit Fab (VEGFR, 111-116-144; Jackson Immunoresearch, Baltimore, PA, USA) for 30 min on ice, respectively. After incubation cells were washed with PBS-BSA 1% (w/v). Data were acquired using an FACScalibur cytometer (Becton Dickinson, Heidelberg, Germany) equipped with a 488-nm argon laser and a 635-nm red diode laser and analyzed using CellQuest software (Becton Dickinson, San Jose, CA, USA). The setup of FACScalibur was performed according to the manufacturer’s instructions using unstained and single-antibody stained cells. Specificity of staining was controlled by incubation with isotype-matched immunoglobulins. To quantify total peripheral endothelial cells, the numbers of KDR-positive cells, to quantify EPCs, the numbers of CD133/KDR double-positive cells within the myelomonocytic cell population were counted.
Colony-forming unit (CFU) assay
The assay was performed by using the EndoCult Liquid Medium Kit® (StemCell Technologies, Vancouver, BC, Canada) per the manufacturer’s protocol. MNCs were resuspended in complete EndoCult medium and seeded at 5 × 106 cells/well on fibronectin-coated tissue culture plates (BD Biosciences, Rockville, MD, USA). After 48 h, wells were washed with media and non-adherent cells were collected. Non-adherent cells were plated in their existing media at 106 cells/well in 24-well fibronectin-coated tissue culture plates for 3 days. Only colonies with at least 20 cells, containing rounded cells in the middle and elongated cells at the periphery, were considered as CFU-EC colonies. The numbers of colonies (colonies/well) appearing after this period were counted. At least two members of the laboratory staff evaluated the numbers of CFU-ECs. They were blinded for the diagnosis and status of the investigated patients/controls. In all patients, the phenotype of cells within the colonies was determined more in detail. For this purpose, cells were characterized by the uptake of DiI-labeled acetylated low-density lipoprotein (ac-LDL) (Invitrogen, Carlsbad, CA, USA) and binding of FITC-labeled UE lectin (Sigma Diagnostics, St. Louis, MO, USA). Cells were first incubated with 10 µg/mL DiI-ac-LDL at 37 °C for 1 h and later fixed with 2% formaldehyde for 10 min, followed by incubation with UE lectin at 37 °C for 1 h. The number of Dil-acLDL+/UE lectin+ cells was counted by laser scanning microscopy using an inverted fluorescence microscope IX-71 (Olympus Deutschland GmbH, Hamburg, Germany) equipped with the appropriate excitation and emission filters (AHF Analysentechnik, Tuebingen, Germany).
Enzyme-linked immunosorbent assay (ELISA)
Serum concentrations of the following vasomodulatory mediators were quantified by ELISA technique according to the manufacturer’s protocols: vascular endothelial growth factor (VEGF), angiopoietin-1 and -2 (Ang-1/Ang-2), platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-b) (all from R&D, Wiesbaden, Germany).
Statistical analysis
Data were analyzed for normality using the Shapiro-Wilk test. Differences between two groups were analyzed by the Mann-Whitney test. Correlation analyses were performed using the Spearman’s rank correlation test. Significance was postulated if P-values were below 0.05. Data are given as mean ± standard deviation (SD) or (follow-up analyses) as mean ± standard error of the mean (SEM).
| Results | ▴Top |
Patients
A total number of 50 individuals were included in the study (58% males and 42% females). The mean age of all subjects was 45 ± 15 years. Seventy-four percent of the patients were HLA-B27-positive. In 38%, at least one relative of the first or second degree suffered from AS. Thirty-three subjects (66%) presented with AS, and the remaining 17 individuals (34%) were diagnosed with nr(non-radiographic)-axSpA. The mean duration of the disease was 9 ± 9 years. Mean CRP and mean erythrocyte sedimentation rate (ESR) at the time of diagnosis were 8.1 ± 18.7 mg/L and 11/21 ± 18/23 mm. Thus, 42% of the patients showed laboratory findings of humoral inflammation. Physiotherapy was performed on a regular basis in 30 of 50 subjects (60%), and the mean duration of morning stiffness was 32.6 ± 35.4 min. Fifty percent of the subjects (n = 25) showed at least one or more co-morbidities: 32% suffered from arterial hypertension, and the mean body mass index was 26.1 ± 5.0 kg/m2, thus ranging within the “overweight” but not “obesity” section according to recently published criteria [24]. The prevalence of diabetes mellitus was low (4%, n = 2). Thirty-eight percent reported a history of smoking, and the mean number of pack-years was 12. Therapy with non-steroidal anti-inflammatory drugs (NSAIDs) was performed in 82%, with 71% on a regular and 29% on an intermittent basis. Proton pump inhibitors (PPIs) were applied in 54% and statins in 8%. Sulfasalazine was administered in 30% and 32 patients (64%) received anti-TNF-alpha inhibitors (adalimumab or etanercept or infliximab). Table 1 summarizes the patients’ characteristics.
 Click to view | Table 1. Patient’s Baseline Characteristics |
EPC colony formation
Regenerative EPC activity was evaluated by using an established CFU assay. In SpA, numbers of colonies were significantly lower as compared to untreated controls (25 ± 20 vs. 37 ± 25.6, P = 0.02).
We next aimed to analyze colony formation in certain subcategories, with all of these assigned to one of two groups, respectively. The first group contains disease-related subcategories (e.g. humoral inflammation, disease activity and patients’ mobility), and the second group summarizes different CVR factors.
Disease-related subcategories
There was no difference in colony numbers between HLA-B27-positive and negative patients (HLA-B27+: 25 ± 21.5 vs. HLA-B27-: 26 ± 13.6, P = 0.52). Next, several disease activity-related parameters were evaluated. Disease activity was quantified using the BASDAI. Patients with more disease activity tended to have higher numbers of colonies than those with less activity but the difference was not statistically significant. Nevertheless, patients with less activity showed fewer numbers of colonies as compared to the controls (21 ± 19 vs. 37 ± 25.6, P = 0.001). There was no difference between patients with BASDAI < 4 as compared to those with higher values. Additional analyses were performed using the ASDAS. Patients with low activity showed significantly fewer colonies than healthy controls (16 ± 6.6 vs. 37 ± 25.6, P = 0.02). Individual mobility was measured by the BASMI, the BASFI, the tests of Schober and Ott, and the FFD, respectively. None of the parameters correlated with colony formation (P-values: BASMI 0.37, BASFI 0.86, Schober 0.68, Ott 0.15, FFD 0.89).
Humoral inflammation was quantified by measurements of the CRP. Patients with a value of < 5 mg/dL were defined as those with mild to moderate humoral inflammatory activity, and patients with a value of > 5 mg/dL were defined as highly active. The difference in colony numbers was not significant (21 ± 15 vs. 19 ± 19.7, P = 0.58). The individual pharmacological treatment regimens significantly differed between axSpA patients. Several drugs have been shown to modulate EPC regeneration and numbers of blood-derived EPCs. We were particularly interested in NSAIDs, TNF-alpha neutralizing agents and PPIs. We did not find any difference between patients receiving these types of drugs and healthy controls in terms of colony formation (NSAIDs+ vs. NSAIDs- and controls: 24.7 ± 17.9 vs. 28 ± 30 and 37 ± 25.6, P = 0.69 and 0.9; axSpA receiving anti-TNF-alpha vs. axSpA without anti-TNF-alpha: 27.8 ± 20.6 vs. 22 ± 16.1, P = 0.27; axSpA receiving PPI vs. axSpA without PPI: 26 ± 22 vs. 24 ± 18.5, P = 0.4). Figure 1 summarizes the results of all disease-related subcategories.
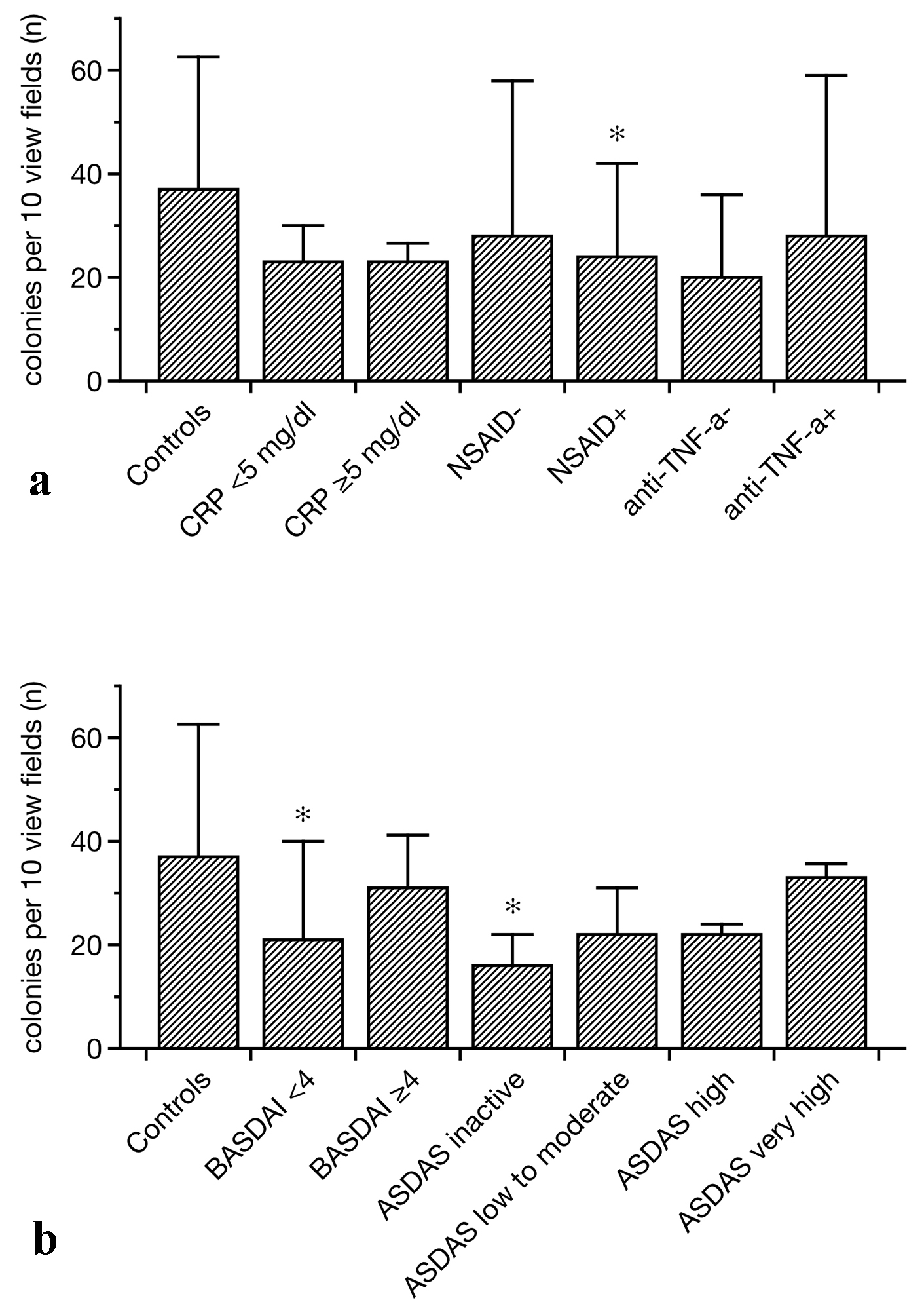 Click for large image | Figure 1. EPC colony formation in controls and SpA patients related to different treatments (a) and disease-associated categories (b). In general, colony numbers were lower in SpA as compared to controls. Particularly patients with a BASDAI of lower than 4 and individuals with the lowest ASDAS displayed impaired EPC colony formation (b). In addition, NSAID treatment was associated with lower than normal colony formation (data as median ± lower and upper quartile, *P < 0.05 as compared to the controls). EPCs: endothelial progenitor cells; SpA: spondylarthritis; BASDAI: Bath ankylosing spondylitis disease activity index; ASDAS: Ankylosing spondylitis disease activity score; NSAIDs: non-steroidal anti-inflammatory drugs. |
CRV subcategories
Colony numbers did not correlate with age at the time of inclusion into the study (P = 0.23). Females displayed more colonies than males (female 32 ± 7 vs. male 20 ± 6 colonies, P = 0.003). Since obesity, arterial hypertension and smoking are well-known CVR factors, we aimed to analyze these parameters in relation to the numbers of EPC colonies. The body mass index (BMI) did not correlate with colony formation (P = 0.7), but axSpA patients with arterial hypertension showed significantly fewer colonies than healthy controls (18.9 ± 14 vs. 37 ± 25.6, P = 0.007). The axSpA patients with ongoing nicotine abuse did not show less colonies than non-smoking axSpA patients but displayed fewer colonies than healthy controls (20 ± 17 vs. 27 ± 24 and 37 ± 25.6, P = 0.4 and 0.005). Colony numbers tended to be lower in non-smoking axSpA patients than in controls (27 ± 24 vs. 37 ± 25.6, P = 0.07). Patients undergoing statin treatment did not show higher colony number than those without therapy (no statins 25 ± 20, statins 26 ± 22, P = 0.93). Finally, we evaluated the impact of regularly performed physical training but once again, we failed to detect any difference (no exercise 23 ± 15, exercise 26 ± 22, P = 0.52). Figure 2 summarizes the results of all CVR subcategories.
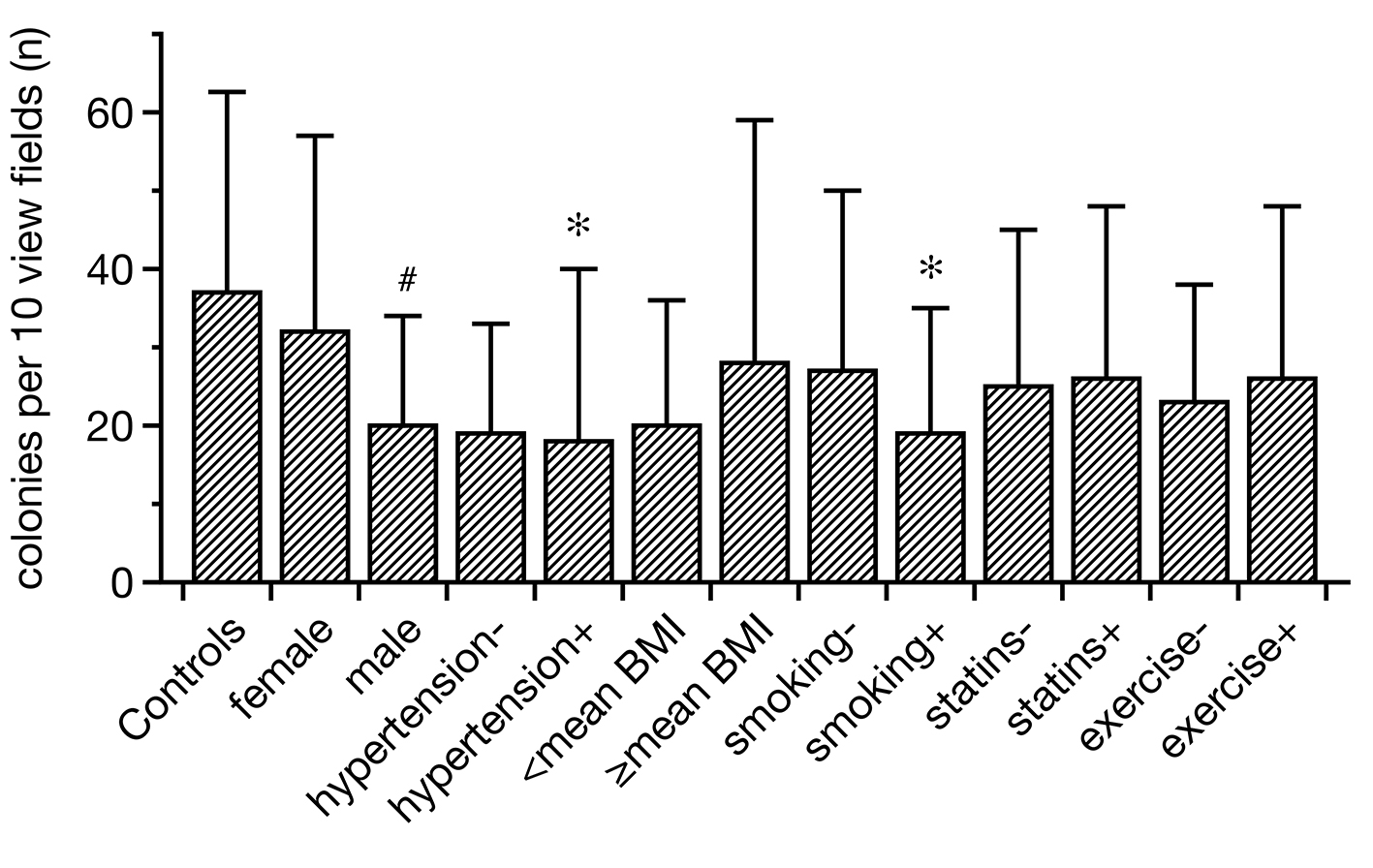 Click for large image | Figure 2. Regenerative activity of EPCs in controls and SpA related to certain CVR factors. Hypertensive and smoking SpA patients displayed lower EPC colonies than healthy subjects. In addition, males showed less intense colony formation than females. All other categories did not differ from each other/the controls in a significant manner (data as median ± lower and upper quartile, *P < 0.05 as compared to the controls; #P < 0.05 between females and males). EPCs: endothelial progenitor cells; SpA: spondylarthritis; CVR: cardiovascular risk. |
Peripheral circulating EPCs
In addition to colony formation analysis, we also enumerated peripheral circulating EPCs as published in previous studies [19, 20, 23]. It needs nevertheless to be mentioned that a specific set of EPC markers is still missing. Thus, the combined staining of CD133 and KDR is an approximative method for EPC quantification at least. Peripheral CD133+/KDR+ cells did not differ between controls and axSpA patients (controls vs. SpA: 0.75±0.85% vs. 0.97±1.65%, not significant). Thus, we did not perform further analyses related to circulating cell numbers at this point.
Follow-up
A total number of 34 out of 50 included patients (68%) were additionally followed up after a mean period of 2 years after inclusion into the study. The following activity- and damage-associated parameters were re-evaluated: BASDAI, Ott’s sign (cm) and FFD (cm). Differences (Δ-values) to the initial values were calculated and related to gender, EPC colony numbers, peripheral circulating EPCs and serum levels of VEGF, Ang-1/Ang-2, PDGF and TGF-b at the time of study inclusion. In all categories except gender, the respective mean values were used as cut-off for comparisons. Differences were not statistically significant in any of the analyzed subgroups: female - male/below vs. ≥ mean number of colonies (CFU-ECs)/below vs. ≥ mean percentages of CD133+/KDR+ cells/below vs. ≥ mean serum levels of VEGF/Ang-1/Ang-2/PDGF/TGF-b (Fig. 3).
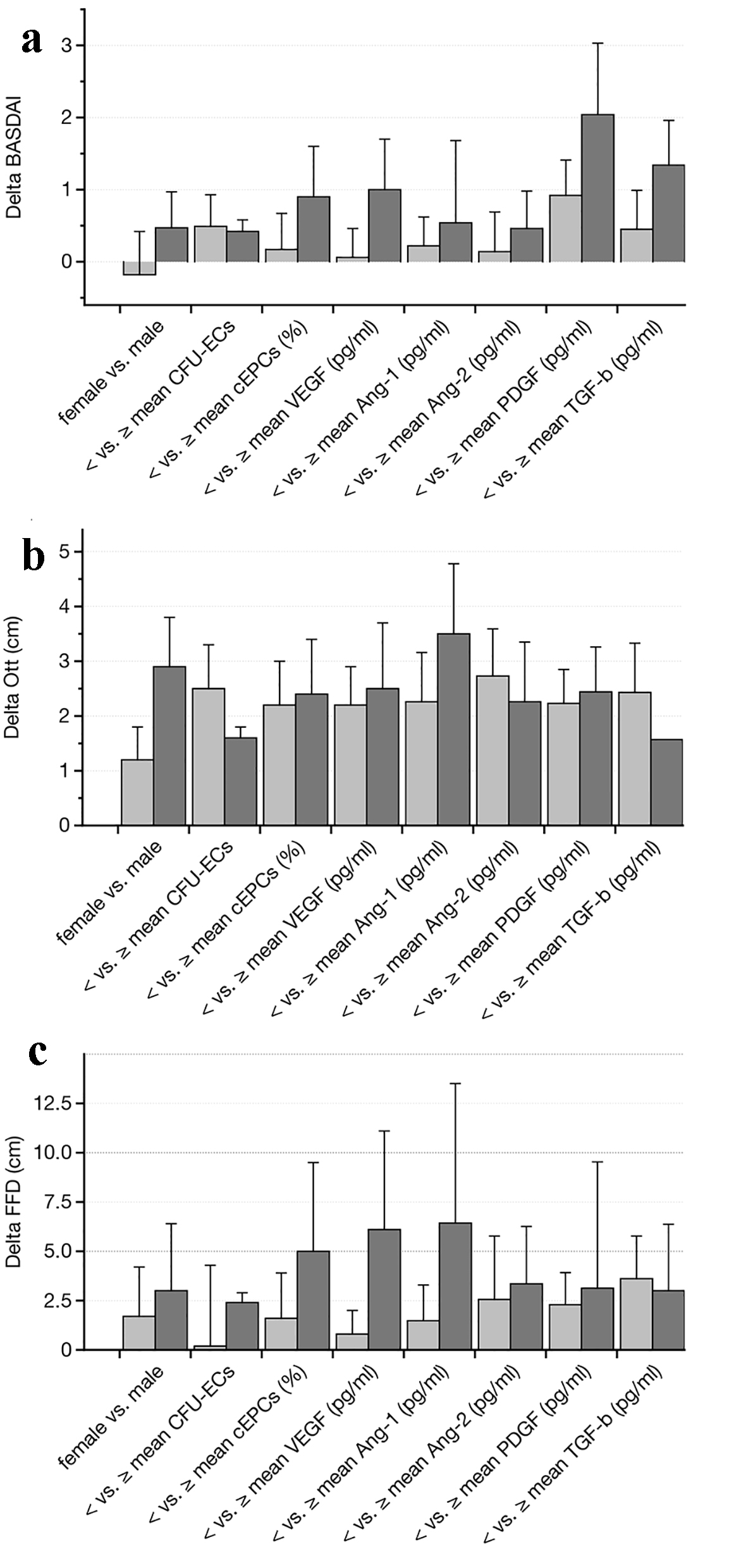 Click for large image | Figure 3. Follow-up analysis after a mean period of 2 years (performed in 34 out of 50 patients (68%)). The parameters including gender, mean number of EPC colonies, peripheral circulating EPCs, and mean serum levels of VEGF, angiopoietin-1 and -2, PDGF and TGF-b were used for comparing differences in BASDAI (a), Ott’ sign (b) and FFD (c) (both in cm) over time. None of the analyzed subgroups showed any significant difference as compared to the beginning of the study (data as median ± lower and upper quartile). EPCs: endothelial progenitor cells; BASDAI: Bath ankylosing spondylitis disease activity index; VEGF: vascular endothelial growth factor; PDGF: platelet-derived growth factor; TGF-b: transforming growth factor-beta; FFD: finger-floor distance. |
| Discussion | ▴Top |
The current study is the very first investigation of the eEPC system in axSpA. It is in addition one of the first studies that evaluated EPCs in relation to disease-associated parameters and to CVR factors in SpA in general. Few other groups published data about the cells in other types of SpA such as psoriatic and enteropathic arthritis [25-27]. The title of a more recent study performed in AS suggested that EPCs were enumerated by flow cytometry [21]. In reality, it can seriously be doubted whether the cell surface maker combination used in the study (CD34 and CD133 combined) truly allowed detecting EPCs. To our understanding, the authors did most likely analyze immature hematopoietic but not EPCs.
Our study in contrast offers several new perspectives on EPCs in rheumatic diseases. Firstly, axSpA was associated with decreased EPC colony formation in general. Secondly, especially lower disease activity, as reflected by a BASDAI of < 4 and by an ASDAS of < 1.3 went in parallel with fewer numbers of blood-derived EPC colonies. In addition, sxSpA patients with arterial hypertension and those with ongoing nicotine abuse (smoking) displayed impaired EPC colony formation. There were no differences in the percentages of circulating CD133+/KDR+ cells between any of the groups. Finally, neither colony numbers nor CD133+/KDR+ cells served as reliable tools for predicting disease activity or functional impairment over a mean period of approximately 30 months.
The data on EPC in SpA are very limited. One study performed in psoriatic arthritis patients has been published in 2009 [27]. Individuals with arthritis did not show higher or lower percentages of CD133+/KDR+ cells as compared to those without arthritis or as compared to healthy controls. Nevertheless, the authors found correlations between cell numbers and VEGF levels/the BASDAI, concluding that EPCs may not essentially be involved in the pathogenesis of the disease. Other rheumatic disorders have been investigated more in detail, particularly rheumatoid arthritis, typically characterized by increased formation of highly vascularized synovial tissue [28-30]. One investigation performed by Park and colleagues showed increased formation of bone erosions to be associated with lower EPC numbers, indicating that adequate cell quantities may be required to maintain the structural integrity within and around joints [31]. Among other rheumatic diseases, EPC-related investigations were performed in systemic lupus erythematosus (SLE) [8, 23], granulomatous polyangiitis (GPA) [19] and systemic sclerosis [32, 33], respectively.
The current study revealed impaired EPC colony formation in axSpA with a more pronounced suppression in patients with low disease activity. These data are somehow difficult to interpret. Following the study by Park et al [31], one may speculate that inhibited EPC regeneration potentially perpetuates joint inflammation. In axSpA as opposed to RA, synovial thickening/proliferation occurs less frequent or intense. The so-called pannus is a hallmark of rheumatoid arthritis (RA). In this situation, it appears plausible that suppressed eEPC regeneration accompanies or possibly even augments inflammatory joint and bone damage. In axSpA in contrast, colony numbers were particularly reduced in individuals with low disease activity. This finding does not fit into our proposed concept of EPC regeneration failure as “pro-inflammatory signal”. Therefore, any reasonable explanation is lacking at the moment.
In other disorders such as chronic kidney disease (CKD), (sub)acute kidney injury (AKI) and systemic sclerosis, severe microvasculopathy has been documented as a hallmark of persistent tissue damage [8, 34]. In these situations, vascular rarefication and occlusion go in parallel with increased fibrogenesis. Impaired EPC colony formation has at least been found in patients with CKD and in septic AKI [18, 20]. We did not particularly assess the degree of radiolographic axSpA progression in our study. Therefore, any definite conclusion about EPC regeneration in relation to the stage of axSpA must be drawn after further, expanded studies.
The second point of interest was the impact of CVR factors on EPC proliferation and quantities. Reduced colony formation was detected in male as compared to female axSpA patients and in hypertensive and smoking individuals in contrast to those without arterial hypertension and nicotine abuse. In the two latter categories, differences were significant in comparison to healthy controls but not within the axSpA group. Other CVR factors did not show any impact on colony formation or circulating EPCs, neither if compared to the controls nor if exclusively analyzed between axSpA patients. These data are not truly surprising since comparable observations have been made in the past, at least in hypertension and nicotine abuse [35, 36]. Statins have been documented to increase peripheral EPCs [37]. Our study failed to confirm these findings, possibly due to the low number of patients undergoing statin treatment (8%). At the moment, we must therefore conclude that the mere presence of axSpA does at least not aggravate findings that were previously reported in healthy, non-rheumatic subjects.
The last section of our analyses was performed in order to evaluate the predictive role of certain soluble vasomodulatory substances in terms of disease activity and patients’ mobility in the long-term (mean follow-up period of 33.2 ± 1.9 months). None of the included parameters (Ang-1 and -2, VEGF, PDGF and TGF-b) did significantly predict changes in the mean BASDAI or the FFD or the Ott’s sign. Even the number of colonies in culture failed to differentiate between these categories. Braun and colleagues reported serum VEGF in AS to be not predictive for radiographic progression of the disease [38]. Only few data are available on serum angiopoietins and SpA. Genre et al found associations between serum osteopontin and Ang-2 in TNF-alpha inhibitor treated AS, but long-term analyzes of progression-associated parameters were not performed.
Conclusions
EPC regeneration is significantly affected in axSpA, whereas percentages of peripheral circulating cells do not differ from healthy controls. The exact reasons for particularly low regenerative activity in individuals with low disease activity remain speculative at the moment.
It needs to be elucidated to which extent impaired EPC colony formation possibly correlates with the disease progression or not.
Finally, axSpA is at least not associated with any significant modulation of CVR factors as compared to non-rheumatic subjects. On the other hand, EPC-related parameters may not be particularly useful for CVR assessment in axSpA.
Acknowledgments
We thank Katrin Schwarze for occasionally assisting in performing the CFU-EC assay.
Financial Disclosure
The study was supported by the Heidenreich von-Siebold Programm (Susann Patschan).
Conflict of Interest
The authors state that they have no conflict of interest.
Informed Consent
Written consent was obtained.
Author Contributions
SP designed the study and recruited all patients. MV performed all colony assay and cytometric analyses. DB performed follow-up analyses. EH assisted in cell analyses (CFU-EC assay and flow cytometry). GAM corrected the manuscript and supplied all experimental equipment. CB assisted in writing the manuscript. DP analyzed data and wrote the manuscript.
Abbreviations
Ang-1/Ang-2: angiopoietin-1 and -2; anti-TNF-alpha: anti-tumor necrosis factor-alpha; AS: ankylosing spondylitis; ASAS: assessment of spondylarthritis; ASDAS: ankylosing spondylitis disease activity score; BASFI: Bath ankylosing spondylitis functional index; BASMI: Bath ankylosing spondylitis metrology index; CASPAR: The ClASsification for Psoriatic Arthritis; CFU: colony-forming unit; EPCs: endothelial progenitor cells; eEPCs: early endothelial progenitor cells; FFD: finger-floor distance; lEPCs: late endothelial progenitor cells; NSAIDs: non-steroidal anti-inflammatory drugs; PDGF: platelet-derived growth factor; PPI: proton-pump inhibitors; axSpA: axial spondylarthritis; nr-axSpA: non-radiographic axSpA; TGF-b: transforming growth factor-beta; VEGF: vascular endothelial growth factor
| References | ▴Top |
- Papagoras C, Voulgari PV, Drosos AA. Atherosclerosis and cardiovascular disease in the spondyloarthritides, particularly ankylosing spondylitis and psoriatic arthritis. Clin Exp Rheumatol. 2013;31(4):612-620.
- Gherghe AM, Dougados M, Combe B, Landewe R, Mihai C, Berenbaum F, Mariette X, et al. Cardiovascular and selected comorbidities in early arthritis and early spondyloarthritis, a comparative study: results from the ESPOIR and DESIR cohorts. RMD Open. 2015;1(1):e000128.
doi pubmed - Skare TL, Verceze GC, Oliveira AA, Perreto S. Carotid intima-media thickness in spondyloarthritis patients. Sao Paulo Med J. 2013;131(2):100-105.
doi pubmed - Poddubnyi DA, Rebrov AP. [Endothelial dysfunction in patients with Bechterew's disease (ankylosing spondylitis)]. Klin Med (Mosk). 2007;85(7):66-69.
- Kang KY, Her YH, Ju JH, Hong YS, Park SH. Radiographic progression is associated with increased cardiovascular risk in patients with axial spondyloarthritis. Mod Rheumatol. 2016;26(4):601-606.
doi pubmed - Pedersen SJ, Sorensen IJ, Lambert RG, Hermann KG, Garnero P, Johansen JS, Madsen OR, et al. Radiographic progression is associated with resolution of systemic inflammation in patients with axial spondylarthritis treated with tumor necrosis factor alpha inhibitors: a study of radiographic progression, inflammation on magnetic resonance imaging, and circulating biomarkers of inflammation, angiogenesis, and cartilage and bone turnover. Arthritis Rheum. 2011;63(12):3789-3800.
doi pubmed - Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964-967.
doi pubmed - Patschan D, Loddenkemper K, Buttgereit F. Molecular mechanisms of glucocorticoid-induced osteoporosis. Bone. 2001;29(6):498-505.
doi - Case J, Mead LE, Bessler WK, Prater D, White HA, Saadatzadeh MR, Bhavsar JR, et al. Human CD34+AC133+VEGFR-2+ cells are not endothelial progenitor cells but distinct, primitive hematopoietic progenitors. Exp Hematol. 2007;35(7):1109-1118.
doi pubmed - Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28(9):1584-1595.
doi pubmed - Richardson MR, Yoder MC. Endothelial progenitor cells: quo vadis? J Mol Cell Cardiol. 2011;50(2):266-272.
doi pubmed - Hecht N, Schneider UC, Czabanka M, Vinci M, Hatzopoulos AK, Vajkoczy P, Woitzik J. Endothelial progenitor cells augment collateralization and hemodynamic rescue in a model of chronic cerebral ischemia. J Cereb Blood Flow Metab. 2014;34(8):1297-1305.
doi pubmed - Huang H, Huang F, Huang JP. Transplantation of bone marrowderived endothelial progenitor cells overexpressing Deltalike4 enhances functional neovascularization in ischemic myocardium. Mol Med Rep. 2013;8(5):1556-1562.
doi pubmed - MacArthur JW, Jr., Purcell BP, Shudo Y, Cohen JE, Fairman A, Trubelja A, Patel J, et al. Sustained release of engineered stromal cell-derived factor 1-alpha from injectable hydrogels effectively recruits endothelial progenitor cells and preserves ventricular function after myocardial infarction. Circulation. 2013;128(11 Suppl 1):S79-86.
doi pubmed - Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC, Sordi A, et al. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012;82(4):412-427.
doi pubmed - Patschan D, Kribben A, Muller GA. Postischemic microvasculopathy and endothelial progenitor cell-based therapy in ischemic AKI: update and perspectives. Am J Physiol Renal Physiol. 2016;311(2):F382-394.
doi pubmed - Chan KH, Simpson PJ, Yong AS, Dunn LL, Chawantanpipat C, Hsu C, Yu Y, et al. The relationship between endothelial progenitor cell populations and epicardial and microvascular coronary disease-a cellular, angiographic and physiologic study. PLoS One. 2014;9(4):e93980.
doi pubmed - Bahlmann FH, DeGroot K, Duckert T, Niemczyk E, Bahlmann E, Boehm SM, Haller H, et al. Endothelial progenitor cell proliferation and differentiation is regulated by erythropoietin. Kidney Int. 2003;64(5):1648-1652.
doi pubmed - Patschan S, Patschan D, Henze E, Blaschke S, Wessels JT, Muller GA. Impairment and Differential Expression of PR3 and MPO on Peripheral Myelomonocytic Cells with Endothelial Properties in Granulomatosis with Polyangiitis. Int J Nephrol. 2012;2012:715049.
doi pubmed - Patschan SA, Patschan D, Temme J, Korsten P, Wessels JT, Koziolek M, Henze E, et al. Endothelial progenitor cells (EPC) in sepsis with acute renal dysfunction (ARD). Crit Care. 2011;15(2):R94.
doi pubmed - Verma I, Syngle A, Krishan P. Endothelial progenitor cell biology in ankylosing spondylitis. Int J Rheum Dis. 2015;18(3):336-340.
doi pubmed - Rudwaleit M, van der Heijde D, Landewe R, Akkoc N, Brandt J, Chou CT, Dougados M, et al. The Assessment of SpondyloArthritis International Society classification criteria for peripheral spondyloarthritis and for spondyloarthritis in general. Ann Rheum Dis. 2011;70(1):25-31.
doi pubmed - Patschan S, Patschan D, Potulski M, Henze E, Scholze J, Muller GA. Endothelial progenitor cells in systemic lupus erythematosus. J Nephrol. 2013;26(6):1065-1072.
doi pubmed - Komaroff M. For Researchers on Obesity: Historical Review of Extra Body Weight Definitions. J Obes. 2016;2016:2460285.
doi pubmed - Batycka-Baran A, Paprocka M, Baran W, Szepietowski JC. Decreased number of circulating endothelial progenitor cells (CD133+/KDR+) in patients with psoriatic arthritis. Acta Derm Venereol. 2016;96(6):754-757.
- Bandinelli F, Milia AF, Manetti M, Lastraioli E, M DA, Tonelli P, Fazi M, et al. Lymphatic endothelial progenitor cells and vascular endothelial growth factor-C in spondyloarthritis and Crohn's disease: two overlapping diseases? Clin Exp Rheumatol. 2015;33(2):195-200.
- Ablin JN, Goldstein Z, Aloush V, Matz H, Elkayam O, Caspi D, Swartzenberg S, et al. Normal levels and function of endothelial progenitor cells in patients with psoriatic arthritis. Rheumatol Int. 2009;29(3):257-262.
doi pubmed - Ablin JN, Boguslavski V, Aloush V, Elkayam O, Paran D, Caspi D, George J. Effect of anti-TNFalpha treatment on circulating endothelial progenitor cells (EPCs) in rheumatoid arthritis. Life Sci. 2006;79(25):2364-2369.
doi pubmed - Herbrig K, Haensel S, Oelschlaegel U, Pistrosch F, Foerster S, Passauer J. Endothelial dysfunction in patients with rheumatoid arthritis is associated with a reduced number and impaired function of endothelial progenitor cells. Ann Rheum Dis. 2006;65(2):157-163.
doi pubmed - Yiu KH, Wang S, Mok MY, Ooi GC, Khong PL, Lau CP, Lai WH, et al. Role of circulating endothelial progenitor cells in patients with rheumatoid arthritis with coronary calcification. J Rheumatol. 2010;37(3):529-535.
doi pubmed - Park YJ, Kim JY, Park J, Choi JJ, Kim WU, Cho CS. Bone erosion is associated with reduction of circulating endothelial progenitor cells and endothelial dysfunction in rheumatoid arthritis. Arthritis Rheumatol. 2014;66(6):1450-1460.
doi pubmed - Kuwana M, Okazaki Y. Brief report: impaired in vivo neovascularization capacity of endothelial progenitor cells in patients with systemic sclerosis. Arthritis Rheumatol. 2014;66(5):1300-1305.
doi pubmed - Mok MY, Yiu KH, Wong CY, Qiuwaxi J, Lai WH, Wong WS, Tse HF, et al. Low circulating level of CD133+KDR+cells in patients with systemic sclerosis. Clin Exp Rheumatol. 2010;28(5 Suppl 62):S19-25.
- Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, et al. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol. 2011;300(3):F721-733.
doi pubmed - Li W, Du DY, Liu Y, Jiang F, Zhang P, Li YT. Long-term nicotine exposure induces dysfunction of mouse endothelial progenitor cells. Exp Ther Med. 2017;13(1):85-90.
doi pubmed - Luo S, Xia W, Chen C, Robinson EA, Tao J. Endothelial progenitor cells and hypertension: current concepts and future implications. Clin Sci (Lond). 2016;130(22):2029-2042.
doi pubmed - Urbich C, Dimmeler S. Risk factors for coronary artery disease, circulating endothelial progenitor cells, and the role of HMG-CoA reductase inhibitors. Kidney Int. 2005;67(5):1672-1676.
doi pubmed - Braun J, Baraliakos X, Hermann KG, Xu S, Hsu B. Serum vascular endothelial growth factor levels lack predictive value in patients with active ankylosing spondylitis treated with golimumab. J Rheumatol. 2016;43(5):901-906.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.