

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://www.jocmr.org |
Review
Volume 13, Number 2, February 2021, pages 75-81
Isolated Type Immunoglobulin G4 Sclerosing Cholangitis: The Misdiagnosed Cholangiocarcinoma
Roni Nassera, Hayim Gilshteinb, Subhi Mansourc, Kamel Yasina, Giuseppe Borzellinod, Safi Khuric, e
aGastroenterology and Hepatology Department, Rambam Health Care Campus, Haifa, Israel
bColorectal Surgery Unit, General Surgery Department, Rambam Health Care Campus, Haifa, Israel
cHPB and Surgical Oncology Unit, General Surgery Department, Rambam Health Care Campus, Haifa, Israel
dGeneral Surgery Department, University hospital of Verona, Verona, Italy
eCorresponding Author: Safi Khuri, HPB and Surgical Oncology Unit, General Surgery Department, Rambam Health Care Campus, Haa’leya Hashniya, Haifa 31096, Israel
Manuscript submitted January 14, 2021, accepted January 21, 2021, published online February 25, 2021
Short title: IgG4-Related Sclerosing Cholangitis
doi: https://doi.org/10.14740/jocmr4428
- Abstract
- Introduction
- Methods
- Historical Perspectives and Diagnostic Criteria
- Isolated Type IgG4-SC vs. Cholangiocarcinoma
- Clinical Presentation
- Serological Tests
- Radiological Differences and Histopathological Findings
- Summary
- References
| Abstract | ▴Top |
Immunoglobulin G4 sclerosing cholangitis (IgG4-SC), firstly described in 2004, is the biliary manifestation of a recently described multisystem immune-mediated disease known as IgG4-related disease. IgG4-SC is a unique and rare type of cholangitis of unknown etiology and its precise prevalence rate is still unclear. It is characterized by bile duct wall thickening and high levels of systemic serum IgG4 plasma cells. Differential diagnoses for IgG4-SC include benign (primary sclerosing cholangitis) as well as malignant (extra-hepatic cholangiocarcinoma) diseases. Discrimination between these entities is very important, due to the fact that they have different biological behaviors and different therapeutic strategies. The rare IgG4-SC subgroup with its puzzling manifestations carries a hefty diagnostic challenge for the treating physicians, and inaccurate diagnosis can lead to unnecessary morbid surgical procedures. With the paucity and relative weakness of available data in the current literature, one needs to carefully review all available parameters. A low threshold of suspicion is required to try and prevent missing IgG4-SC. IgG4-SC is highly responsive to steroid treatment, especially during the early inflammatory phase, while delay in management could lead to fibrosis and organ dysfunction. On the other hand, cholangiocarcinoma is treated by means of surgery and/or chemotherapeutic agents.
Keywords: Immunoglobulin G4 sclerosing cholangitis; Extra-hepatic cholangiocarcinoma; Autoimmune pancreatitis
| Introduction | ▴Top |
Immunoglobulin G4-related disease (IgG4-RD), also known as IgG4-related sclerosing disease or IgG4-associated disease, is a recently described systemic disease characterized by inflammation and fibrosis of the affected tissues [1, 2]. Inflammation usually occurs during the early phase of the disease, whereas fibrosis develops later when diagnosis is delayed. Although it is well known as an immune-mediated disease, and there is a growing evidence that the disease is an autoimmune in nature, the precise pathogenesis is yet to be known. It usually results in pseudotumorous swelling of the affected organs, along with high levels of serum IgG4 plasma cells [3, 4]. The affected tissues usually share similar pathological features, with dense lyphoplasmacytic infiltrate rich in IgG4-positive plasma cells being the hallmark findings [2]. Diagnosis of IgG4-RD usually relies on a combination of clinical, imaging, histological and serological findings, as no single one is diagnostic.
The exact prevalence of this disease is unknown due to the fact that it is not well recognized by many practitioners. In Japan, where IgG4-RD is apparently common, the estimated prevalence rate (which is believed to be underestimated) was reported to be 0.28 - 1.08/100,000 population [5]. The growing awareness of the treating physicians for this underestimated entity may result in more accurate epidemiological data. IgG4-RD is more common in males than in females, and usually affects middle-aged and elderly patients with an onset at 50 - 70 years, although rare pediatric cases have been described as well [6]. IgG4-RD can affect any organ system, with the pancreas and salivary glands being the most common affected organs. Other less common organs to be involved include bile ducts, thyroid gland, aorta, retroperitoneum, orbits, lacrimal glands, meninges, lungs and kidneys [1, 2, 7]. Although IgG4-RD can affect single organ, most patients (60-90%) suffer multiple organ involvement at presentation [8, 9]. The American College of Rheumatology and European League Against Rheumatism use the cohort of Wallace et al [10], which included 765 patients with IgG4-RD, to recognize four different groups of clinical phenotypes: hepatopancreatobiliary disease (31%), retroperitoneal fibrosis and/or aortitis (24%), disease limited to the head and neck (24%) and Mikulicz syndrome (lacrimal and parotid gland enlargement) with systemic involvement (22%).
Immunoglobulin G4 sclerosing cholangitis (IgG4-SC), also known as IgG4-associated cholangitis [11], is the biliary system manifestation of the IgG4-RD. IgG4-SC is known as a recent type of sclerosing cholangitis that differs from primary sclerosing cholangitis (PSC). Previously, cholangitis was classified into two types: primary, which is usually idiopathic, and secondary, which could be due to bile duct stones, cholangiocarcinoma, congenital biliary disorders or postoperative bile duct injury [12]. Today, with the emergence of IgG4-SC type, that is the most common type, three types are identified.
IgG4-SC is associated with type 1 autoimmune pancreatitis (AIP-1) (the prototype of IgG4-RD) in more than 90% of cases. Due to this association, they are sometimes collectively called sclerosing pancreatocholangitis. In their study, of 53 patients with IgG4-SC, Ghazale et al [13] demonstrated that only 7.5% suffered from sclerosing cholangitis without AIP. In the absence of associated AIP, it is very challenging to diagnose IgG4-SC. IgG4-related retroperitoneal fibrosis and IgG4-related sialadenitis are also found occasionally in patients suffering from IgG4-SC.
IgG4-SC may present in different manners: either as tumorous mass due to segmental bile duct involvement, which should be differentiated from cholangiocarcinoma, or as a diffuse sclerosing process mimicking PSC [14, 15].
The differentiation between isolated type IgG4-SC and cholangiocarcinoma is both challenging and crucial due to the dissimilar therapeutic strategies for those diagnostic entities. Thus, major morbid procedure can be potentially avoided by making the correct diagnosis.
Objective
The aim was to review the pertinent and available studies in the English literature in order to try and delineate clinical, radiological, serological and histopathological findings that can help to differentiate between isolated type IgG4-SC and cholangiocarcinoma.
| Methods | ▴Top |
A search in PubMed was conducted, based on the PICOS acronym. Headings and text words were used to identify studies that compare isolated type IgG4-SC and cholangiocarcinoma.
The following search terms were included: “IgG4-related disease”, “isolated type IgG4 sclerosing cholangitis”, “IgG4 cholangiopathy”, “isolated type IgG4-SC vs. cholangiocarcinoma”, “isolated type IgG4 cholangiopathy vs. cholangiocarcinoma”, “difference between IgG4-SC and cholangiocarcinoma”.
| Historical Perspectives and Diagnostic Criteria | ▴Top |
IgG4-RD is a disease that has emerged and was studied mainly during the last two to three decades. Although several single organ manifestations of IgG4-RD have been described during the 19th century, such as Mikulicz’s disease (enlargement of the lacrimal and parotid glands) and Riedel’s thyroiditis (a rare type of thyroiditis that presents as a hard goiter), it is in 1995 that the initial hint for the identification of IgG4-RD was made by the identification of steroid responsive autoimmune pancreatitis, known today as AIP-1 (IgG4-related pancreatitis) [16]. Later on, in 2001, Hamano et al [17] found high levels of serum IgG4 plasma cells in patients with sclerosing pancreatitis, treated by corticosteroids to induce clinical improvement and significant decrement of IgG4 concentrations. A year after (2002), he described the multiorgan involvement of IgG-RD when identical histopathological changes were found in concomitant retroperitoneal fibrosis [18].
IgG4-SC, firstly described in 2004, is the most common extra-pancreatic manifestation of AIP-1, developing in 20-88% of cases [19, 20]. Since the introduction of IgG4-SC as a unique entity of cholangitis, different nomenclatures were proposed. In 2009, the European Association for the Study of the Liver (EASL) introduced the nomenclature IgG4-associated cholangitis [21]. This was later changed to IgG4-related sclerosing cholangitis by an organizing committee, composed of 35 experts in IgG4-RD from different countries, during the first international symposium on IgG4-RD [22]. During this symposium, the committee emphasizes the importance of including the word “sclerosing”, although not all bile ducts demonstrate residual sclerosis following steroid management.
Following the introduction of IgG4-RD as a newly diagnosed medical entity, several diagnostic criteria were proposed with large differences in therapeutic strategies between western and eastern countries. The Japanese Pancreatic Society was the first to introduce a diagnostic criterion for AIP based on radiological findings, serological tests and histopathological changes in 2002 and 2006 [23, 24]. It is only in 2006 that IgG4 was added as part of the serological diagnostic findings of the disease. In the same year, Chari et al of the Mayo Clinic introduced the HISORt criteria, composed of five elements, for the diagnosis of AIP [20]. The five elements for diagnosis included: Histological findings, Imaging, Serological tests, Other organ involvement and Response to steroid treatment. Endoscopic retrograde cholangiopancreatography (ERCP), which was used by the Japanese as an important dignostic tool, was excluded according to the HISTORt criteria, due to the risk of post-ERCP pancreatitis as a complication. These criteria were then extended to be used also for the diagnosis of IgG4-SC (Table 1 [20]).
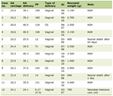 Click to view | Table 1. The HISTORt Criteria for the Diagnosis of IgG4-SC |
In 2010, the International Consensus Diagnostic Criteria was introduced [25]. These criteria depend on the same features of HISTORt, except for steroid responsiveness being optional, as well as introducing the level of evidence for most of the diagnostic factors. IgG4 levels greater than twofold are proposed as level 1 criteria and lesser elevation is level 2. A year later, the Japanese Pancreatic Society developed a revised criterion, with ERCP being an intermediate imaging evidence [23]. In 2012, new clinical diagnostic criteria for IgG4-SC were established by Ohara et al [26]. The aforementioned criteria depend on four main factors: high levels of serum IgG4 plasma cells, biliary imaging features, characteristic histopathological findings and other organ involvement. Steroid responsiveness is optional. Although several diagnostic criteria were developed, the most common in use is the HISORt criteria.
IgG4-SC is usually classified into four types according to site of bile duct involvement on imaging [27]. Type 1 IgG4-SC, the most common type, involves stricture at the distal bile ducts and should be discriminated from cholangiocarcinoma and pancreatic cancer. Type 2 involves diffuse bile duct stenosis through the intra-/extra-hepatic ducts, with a differential diagnosis that includes mainly PSC. Type 3 is characterized by stenosis at the distal bile ducts as well as at the hepatic hilar ducts, while type 4 involves stricture at the hepatic hilar bile ducts (Fig. 1). Of note, cholangiocarcinoma is the main differential diagnosis of all types of IgG4-SC, except type 2.
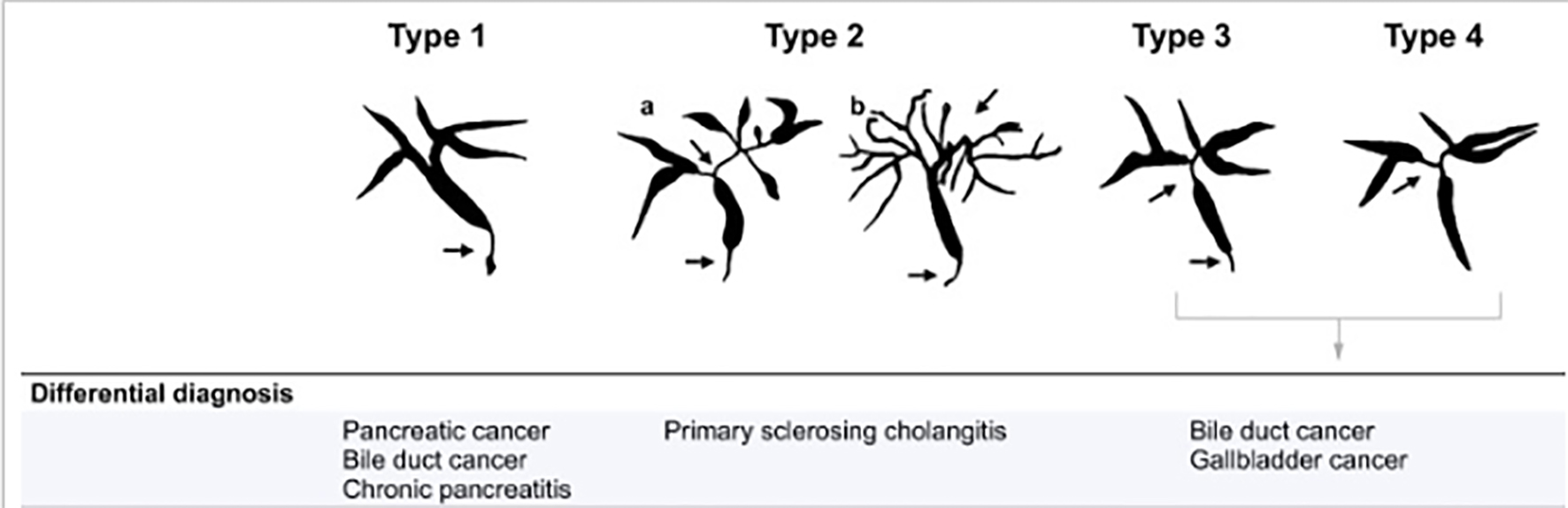 Click for large image | Figure 1. The different types of IgG4-SC with the differential diagnosis. Arrows indicate sites of strictures/stenosis. IgG4-SC: immunoglobulin G4 scelrosing cholangitis. |
| Isolated Type IgG4-SC vs. Cholangiocarcinoma | ▴Top |
Extra-hepatic cholangiocarcinoma is one of the main differential diagnoses of IgG4-SC. Diagnosis of isolated type IgG4-SC is very challenging, and usually misdiagnosed as cholangiocarcinoma. Due to differences in biological behaviors, treatment options and survival, it is of immense importance to differentiate between these diseases.
Reviewing the relevant English literature revealed small number (less than 200) of patients reported with isolated type IgG4-SC in relatively small retrospective series and case reports with a very limited comparison between the two entities.
| Clinical Presentation | ▴Top |
Isolated type IgG4-SC, as well as extra-hepatic cholangiocarcinoma, may have a wide range of presentations such as an incidental abnormality in liver function tests, jaundice, pruritus, abdominal pain or weight loss. Painless jaundice which is the presenting symptom in 90% of patients with extra-hepatic cholangiocarcinoma is far less in patients with isolated type IgG4-SC.
One of the largest retrospective cohort studies was reported by Tanaka [28] and included 527 patients with IgG4-SC, of which 70 patients (13%) had the isolated type. The main presenting symptom was jaundice (35%), followed by the absence (28%) and pruritus (13%). Another study [29] included 21 patients with final pathological diagnosis of isolated type IgG4-SC, who underwent operation due to a presumed diagnosis of cholangiocarcinoma. Their main presenting symptom was jaundice (28%) followed by weight loss (25%). In the same study, jaundice was more prevalent as a chief complaint (86%) for patients with a proven diagnosis of cholangiocarcinoma.
Du et al [30] reported a comparative study, including 30 patients with IgG4-SC (five with isolated type IgG4-SC) and 275 with cholangiocarcinoma. Although jaundice was the most common presenting symptom for both groups with no significant statistical difference, weight loss was more common in patients with IgG4-SC than in patients with cholangiocarcinoma with significant statistical difference.
Several other studies [15, 31-34], including small series of patients with an overall of 22 patients with isolated type IgG4-SC, demonstrated that jaundice was the most common presenting symptom being reported in 16 patients out of 20 (the presenting symptom was not reported in two patients) [34].
Additional case reports [35-38] about IgG4-SC mimicking cholangiocarcinoma also demonstrated jaundice as the most prevalent initial presentation.
In regard to patients’ demographic characteristics, most reported studies [15, 28-38] showed that IgG4-SC, as well as cholangiocarcinoma, affects middle aged to elderly patients (50 - 82 years old), and are more common in males than in females.
Weight loss was reported to be more commonly associated with IgG4-SC [30], a finding that was documented in one study only. Other clinical symptoms were not found to be specific for differentiating purposes (Table 2).
Click to view | Table 2. Clinical Differences Between IgG4-SC and Cholangiocarcinoma |
| Serological Tests | ▴Top |
As obstructive jaundice is the most common presentation for both entities, elevated levels of bilirubin (mainly the conjugated type) are to be expected. However, results from different studies are inconclusive. In the study by Tanaka [28], average levels of total bilirubin were at the upper level of normal (1.2 mg/dL) and accurate levels for the isolated type IgG4-SC were not given. Significantly higher levels of total bilirubin were reported by Roos et al [29] in patients with cholangiocarcinoma. Nevertheless, Du et al [30] showed elevated levels of bilirubin by almost fivefold for both entities, while other studies [15, 31, 35, 37, 38] demonstrated a wide range of total bilirubin levels from normal, up to 19-fold increment. In regard to other liver function test levels, there was no statistically significant difference between the two [30]. Alkaline phophatase (ALKP) was elevated in almost 79% of patients with isolated IgG4-SC [28].
Serum IgG4 level is one of the most important markers used for the diagnosis of IgG4-RD. Although it is used as part of the diagnostic criteria for AIP, with a cutoff level of 135 mg/dL, few studies reported the importance of IgG4 levels with regard to the diagnosis of IgG4-SC. IgG4 levels were used as part of diagnostic criteria for IgG4-SC for the first time in 2011 [34]. Based on this comparative study, 15 patients with IgG4-SC were included, of which only two patients were of the isolated type. High levels of IgG4 were significantly noticed in IgG4-SC patients (85%) than in patients with cholangiocarcinoma (22%). The same results were also documented in other study [28], when about 84.4% of patients with IgG4-SC had a serum IgG4 above the normal range. This level was as high as 90% in another study [39]. Lower rates were documented [32], where out of 10 patients with the isolated type IgG4-SC, seven (70%) had elevated IgG4 levels, ranging between 175 and 797 mg/dL (1.25- to 5.4-fold increase).
The largest cohort study using serum IgG4 to discriminate between IgG4-SC and cholangiocarcinoma was conducted by Oseini et al [40]. In his study, which included 126 patients with cholangiocarcinoma and 50 patients with IgG4-SC (with no analysis of the isolated type), 13.5% of cholangiocarcinoma patients had elevated levels of IgG4, with 3.2% (mainly patients with associated PSC) that had more than twofold increment. They concluded that few patients with cholangiocarcinoma, mainly with PSC, have elevated levels of IgG4, with minority of patients demonstrating levels over twofold the normal range. Thus, high IgG4 levels on their own cannot exclude the diagnosis of cholangiocarcinoma. A cutoff level of twofold is not sufficient to differentiate between the two entities, while a cutoff value of four times the upper normal limit has a 100% specificity for diagnosing IgG4-SC. Hence, increasing the cutoff increases the specificity [41].
Values higher than 207 mg/dL may be useful to completely distinguish IgG4-SC (mainly type 3 and 4) from cholangiocarcinoma (Table 2).
There was no study conducted to evaluate the importance of serum IgG4 levels for the diagnosis of the isolated subtypes, and data regarding this specific issue are based on small series of patients or single case reports. Patients with isolated type IgG4-SC usually do not display marked increase in IgG4 levels as compared to AIP-associated types [42].
The report by Hamano et al [15] included three patients with isolated type IgG4-SC showing modest elevation of serum IgG4 levels (119 - 195 mg/dL).
Thus, diagnosis of IgG4-SC, especially the isolated type, cannot be based solely on high levels of IgG4, which is known to have an overall sensitivity and specificity of 64-90% and 87-93%, respectively [39, 40].
| Radiological Differences and Histopathological Findings | ▴Top |
The different radiological tools used nowadays, such as computed tomography (CT) scan, magnetic resonance imaging (MRI), endoscopic ultrasound (EUS) and intraductal US (IDUS) are very important for the identification of bile ducts abnormalities. Current English literature lack studies about radiological and histopathological findings specific for isolated type IgG4-SC. Thus, most available data are taken from studies about non-specific IgG4-SC. Radiological findings that are usually found in IgG4-SC include: bile duct thickening involving intra- and extra-hepatic bile ducts, concentric bile duct wall thickening extending along the long axis; smooth inner/outer margins and visibility of patent bile duct in the strictures [15, 43, 44].
It remains very challenging to distinguish between IgG4-SC and bile duct malignancy based on imaging features alone. Several studies have tried to look for ancilary findings in order to discriminate between the two [15, 44, 45].
Yata et al [44] compared CT scan findings and found that a lesion involving the intrapancreatic bile duct, smooth outer margins, fully visible lumen, a funnel-shaped proximal bile duct, skip lesions and abnormal pancreatic findings favors IgG4-SC while dual-layered attenuation in all phases was significantly more common in extra-hepatic chollangiocarcinoma with a specifity of more than 80%.
Another study [45] found that the presence of pancreatic abnormalities, including peripancreatic ring, atrophy, abnormal enhancement, or T2 signal intensity, strongly favors a diagnosis of IgG4-SC.
After analyzing 162 MRI images ,Tokala et al [46] suggested that the presence of continuous disease in the bile ducts, gallbladder involvement and single-wall common bile duct thickness greater than 2.5 mm supports a diagnosis of IgG4-SC over PSC. Both could not be differentiated on the basis of location and length of common bile duct stricture.
IgG4-SC primarily involves the intra-, extra-, hilar and perihilar bile ducts. It is characterized by wall thickening caused by transmural marked lymphoplasmacytic infiltration and fibrosis. Eosinophilic infiltration, storiform fibrosis and obliterative phlebitis are commonly identified. Inflitration of IgG4 rich cells (> 10 IgG4-positive cells in high-power field (HPF)) in a biopsy is also characterestic but not specific for IgG4-SC. IgG4 rich cells can also be found in PSC and cholangiocarcinoma.
In IgG4-SC, biliary inflammation occurs transmurally, and epithelial layer usually is not affected. In contrast, cholangiocarcinoma arises from the epithelial layer and grows invasively [43, 47-49].
A consensus published in 2012 proposed that 100 IgG4-positive plasma cells per HPF in surgical specimens and 10 per HPF in biopsy samples are required for diagnosis of IgG4-RD. A ratio of IgG4/IgG-positive cell of 40% is helpful in discriminating IgG4-SC from other forms of lymphoplasmacytic cholangitis [50].
| Summary | ▴Top |
IgG4-RD is a fascinating and relatively new clinical entity that encompasses several medical fields in its realm. The rare IgG4-SC subgroup with its puzzling manifestations carries a hefty diagnostic challenge for the treating physicians. The main difficulty is the differential diagnosis between the benign autoimmune process that can be successfully treated with steroids and cholangiocarcinoma, a malignancy with a dismal prognosis. Inaccurate diagnosis can lead to unnecessary morbid surgical procedures and chemotherapeutic treatments with their ensuing physical and emotional burden on our patients. The diagnostic criteria developed throughout the years can help the vigilant clinician to reach the accurate diagnosis. With the paucity and relative weakness of available data in the current literature, one needs to carefully review all available parameters in four different aspects, creating a “quadruple front” including: clinical presentation, serological tests, imaging and pathological findings. A low threshold of suspicion is required to try and prevent missing IgG4-SC. Further studies are needed, preferably multi-center and prospective in nature, in order to improve the level of evidence and improve our understanding of the disease process, diagnosis and treatment.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors have no conflict of interest to declare.
Author Contributions
Acquisition of the search was made by Roni Nasser and Subhi Mansour. The paper was drafted by Safi Khuri, Kamel Yasin and Hayim Gilshtein. Critical revision and final approval of the published version were done by Giuseppe Borzellino and Safi Khuri. All parties agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385(9976):1460-1471.
doi - Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539-551.
doi pubmed - Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34(12):1812-1819.
doi pubmed - Kasashima S, Zen Y, Kawashima A, Konishi K, Sasaki H, Endo M, Matsumoto Y, et al. Inflammatory abdominal aortic aneurysm: close relationship to IgG4-related periaortitis. Am J Surg Pathol. 2008;32(2):197-204.
doi pubmed - Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, Matsui S, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22(1):1-14.
doi pubmed - Wang L, Zhang P, Zhang X, Lin W, Tang H, Li J, Wang M, et al. Sex disparities in clinical characteristics and prognosis of immunoglobulin G4-related disease: a prospective study of 403 patients. Rheumatology (Oxford). 2019;58(5):820-830.
doi pubmed - Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, Stone JH. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466-2475.
doi pubmed - Okazaki K, Uchida K, Koyabu M, Miyoshi H, Takaoka M. Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. J Gastroenterol. 2011;46(3):277-288.
doi pubmed - Sah RP, Chari ST, Pannala R, Sugumar A, Clain JE, Levy MJ, Pearson RK, et al. Differences in clinical profile and relapse rate of type 1 versus type 2 autoimmune pancreatitis. Gastroenterology. 2010;139(1):140-148; quiz e112-143.
doi pubmed - Wallace ZS, Zhang Y, Perugino CA, Naden R, Choi HK, Stone JH, Committee AEI-RCC. Clinical phenotypes of IgG4-related disease: an analysis of two international cross-sectional cohorts. Ann Rheum Dis. 2019;78(3):406-412.
doi pubmed - Hubers LM, Maillette de Buy Wenniger LJ, Doorenspleet ME, Klarenbeek PL, Verheij J, Rauws EA, van Gulik TM, et al. IgG4-associated cholangitis: a comprehensive review. Clin Rev Allergy Immunol. 2015;48(2-3):198-206.
doi pubmed - Okazaki K, Uchida K, Koyabu M, Miyoshi H, Ikeura T, Takaoka M. IgG4 cholangiopathy: current concept, diagnosis, and pathogenesis. J Hepatol. 2014;61(3):690-695.
doi pubmed - Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, Topazian MD, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134(3):706-715.
doi pubmed - Zen Y, Harada K, Sasaki M, Sato Y, Tsuneyama K, Haratake J, Kurumaya H, et al. IgG4-related sclerosing cholangitis with and without hepatic inflammatory pseudotumor, and sclerosing pancreatitis-associated sclerosing cholangitis: do they belong to a spectrum of sclerosing pancreatitis? Am J Surg Pathol. 2004;28(9):1193-1203.
doi pubmed - Hamano H, Kawa S, Uehara T, Ochi Y, Takayama M, Komatsu K, Muraki T, et al. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc. 2005;62(1):152-157.
doi - Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40(7):1561-1568.
doi pubmed - Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732-738.
doi pubmed - Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, Nakazawa K, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359(9315):1403-1404.
doi - Hamano H, Arakura N, Muraki T, Ozaki Y, Kiyosawa K, Kawa S. Prevalence and distribution of extrapancreatic lesions complicating autoimmune pancreatitis. J Gastroenterol. 2006;41(12):1197-1205.
doi pubmed - Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, Clain JE, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006;4(8):1010-1016; quiz 1934.
doi pubmed - European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51(2):237-267.
doi pubmed - Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, Azumi A, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64(10):3061-3067.
doi pubmed - Okazaki K, Kawa S, Kamisawa T, Ito T, Inui K, Irie H, Nishino T, et al. Amendment of the Japanese Consensus Guidelines for Autoimmune Pancreatitis, 2013 I. Concept and diagnosis of autoimmune pancreatitis. J Gastroenterol. 2014;49(4):567-588.
doi pubmed - Okazaki K, Kawa S, Kamisawa T, Naruse S, Tanaka S, Nishimori I, Ohara H, et al. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol. 2006;41(7):626-631.
doi pubmed - Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, Kim MH, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40(3):352-358.
doi pubmed - Ohara H, Okazaki K, Tsubouchi H, Inui K, Kawa S, Kamisawa T, Tazuma S, et al. Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012. J Hepatobiliary Pancreat Sci. 2012;19(5):536-542.
doi pubmed - Nakazawa T, Ohara H, Sano H, Ando T, Joh T. Schematic classification of sclerosing cholangitis with autoimmune pancreatitis by cholangiography. Pancreas. 2006;32(2):229.
doi pubmed - Tanaka A. IgG4-related sclerosing cholangitis and primary sclerosing cholangitis. Gut Liver. 2019;13(3):300-307.
doi pubmed - Roos E, Hubers LM, Coelen RJS, Doorenspleet ME, de Vries N, Verheij J, Beuers U, et al. IgG4-Associated Cholangitis in Patients Resected for Presumed Perihilar Cholangiocarcinoma: a 30-Year Tertiary Care Experience. Am J Gastroenterol. 2018;113(5):765-772.
doi pubmed - Du S, Liu G, Cheng X, Li Y, Wang Q, Li J, Lu X, et al. Differential diagnosis of immunoglobulin G4-associated cholangitis from cholangiocarcinoma. J Clin Gastroenterol. 2016;50(6):501-505.
doi pubmed - Lin J, Cummings OW, Greenson JK, House MG, Liu X, Nalbantoglu I, Pai R, et al. IgG4-related sclerosing cholangitis in the absence of autoimmune pancreatitis mimicking extrahepatic cholangiocarcinoma. Scand J Gastroenterol. 2015;50(4):447-453.
doi pubmed - Oh HC, Kim MH, Lee KT, Lee JK, Moon SH, Song TJ, Eum J, et al. Clinical clues to suspicion of IgG4-associated sclerosing cholangitis disguised as primary sclerosing cholangitis or hilar cholangiocarcinoma. J Gastroenterol Hepatol. 2010;25(12):1831-1837.
doi pubmed - Lytras D, Kalaitzakis E, Webster GJ, Imber CJ, Amin Z, Rodriguez-Justo M, Pereira SP, et al. Cholangiocarcinoma or IgG4-associated cholangitis: how feasible it is to avoid unnecessary surgical interventions? Ann Surg. 2012;256(6):1059-1067.
doi pubmed - Nakazawa T, Naitoh I, Hayashi K, Okumura F, Miyabe K, Yoshida M, Yamashita H, et al. Diagnostic criteria for IgG4-related sclerosing cholangitis based on cholangiographic classification. J Gastroenterol. 2012;47(1):79-87.
doi pubmed - El-Hadary HF, Dadour NM, Ahmed H, Mo’nes D. IgG4-Related disease mimicking cholangiocarcinoma. Clin Radiol Imaging J. 2018;2(1):000117.
- Mittelstaedt A, Meier PN, Dankoweit-Timpe E, Christ B, Jaehne J. IgG4-related sclerosing cholangitis mimicking hilar cholangiocarcinoma (Klatskin tumor): a case report of a challenging disease and review of the literature. Innov Surg Sci. 2018;3(2):157-163.
doi pubmed - Lin HP, Lin KT, Ho WC, Chen CB, Kuo CY, Lin YC. IgG4-associated cholangitis mimicking cholangiocarcinoma - report of a case. J Intern Med Taiwan. 2013;24:137-141
- Rungsakulkij N, Sornmayura P, Tannaphai P. Isolated IgG4-related sclerosing cholangitis misdiagnosed as malignancy in an area with endemic cholangiocarcinoma: a case report. BMC Surg. 2017;17(1):17.
doi pubmed - Boonstra K, Culver EL, de Buy Wenniger LM, van Heerde MJ, van Erpecum KJ, Poen AC, van Nieuwkerk KM, et al. Serum immunoglobulin G4 and immunoglobulin G1 for distinguishing immunoglobulin G4-associated cholangitis from primary sclerosing cholangitis. Hepatology. 2014;59(5):1954-1963.
doi pubmed - Oseini AM, Chaiteerakij R, Shire AM, Ghazale A, Kaiya J, Moser CD, Aderca I, et al. Utility of serum immunoglobulin G4 in distinguishing immunoglobulin G4-associated cholangitis from cholangiocarcinoma. Hepatology. 2011;54(3):940-948.
doi pubmed - Ohara H, Nakazawa T, Kawa S, Kamisawa T, Shimosegawa T, Uchida K, Hirano K, et al. Establishment of a serum IgG4 cut-off value for the differential diagnosis of IgG4-related sclerosing cholangitis: a Japanese cohort. J Gastroenterol Hepatol. 2013;28(7):1247-1251.
doi pubmed - Alswat K, Al-Harthy N, Mazrani W, Alshumrani G, Jhaveri K, Hirschfield GM. The spectrum of sclerosing cholangitis and the relevance of IgG4 elevations in routine practice. Am J Gastroenterol. 2012;107(1):56-63.
doi pubmed - Kamisawa T, Nakazawa T, Tazuma S, Zen Y, Tanaka A, Ohara H, Muraki T, et al. Clinical practice guidelines for IgG4-related sclerosing cholangitis. J Hepatobiliary Pancreat Sci. 2019;26(1):9-42.
doi pubmed - Yata M, Suzuki K, Furuhashi N, Kawakami K, Kawai Y, Naganawa S. Comparison of the multidetector-row computed tomography findings of IgG4-related sclerosing cholangitis and extrahepatic cholangiocarcinoma. Clin Radiol. 2016;71(3):203-210.
doi pubmed - Gardner CS, Bashir MR, Marin D, Nelson RC, Choudhury KR, Ho LM. Diagnostic performance of imaging criteria for distinguishing autoimmune cholangiopathy from primary sclerosing cholangitis and bile duct malignancy. Abdom Imaging. 2015;40(8):3052-3061.
doi pubmed - Tokala A, Khalili K, Menezes R, Hirschfield G, Jhaveri KS. Comparative MRI analysis of morphologic patterns of bile duct disease in IgG4-related systemic disease versus primary sclerosing cholangitis. AJR Am J Roentgenol. 2014;202(3):536-543.
doi pubmed - Graham RP, Smyrk TC, Chari ST, Takahashi N, Zhang L. Isolated IgG4-related sclerosing cholangitis: a report of 9 cases. Hum Pathol. 2014;45(8):1722-1729.
doi pubmed - Zhang L, Lewis JT, Abraham SC, Smyrk TC, Leung S, Chari ST, Poterucha JJ, et al. IgG4+ plasma cell infiltrates in liver explants with primary sclerosing cholangitis. Am J Surg Pathol. 2010;34(1):88-94.
doi pubmed - Fischer S, Trivedi PJ, Ward S, Greig PD, Therapondos G, Hirschfield GM. Frequency and significance of IgG4 immunohistochemical staining in liver explants from patients with primary sclerosing cholangitis. Int J Exp Pathol. 2014;95(3):209-215.
doi pubmed - Teml A, Schwab M, Hommes DW, Almer S, Lukas M, Feichtenschlager T, Florin T, et al. A systematic survey evaluating 6-thioguanine-related hepatotoxicity in patients with inflammatory bowel disease. Wien Klin Wochenschr. 2007;119(17-18):519-526.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.