

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Short Communication
Volume 11, Number 10, October 2019, pages 720-724
Acute Kidney Injury-Associated Systemic Inflammation Is Aggravated in Insulin-Dependent Diabetes Mellitus
Daniel Patschana, c, Katrin Schwarzeb, Elvira Henzeb, Johanna Charlotte Hoffmannb, Susann Patschana, Oliver Rittera, Gerhard Anton Mullerb
aKlinik fur Kardiologie, Angiologie und Nephrologie, Innere Medizin I, Klinikum Brandenburg, Medizinische Hochschule Brandenburg, Brandenburg, Germany
bKlinik fur Nephrologie und Rheumatologie, Universitatsmedizin Gottingen, Gottingen, Germany
cCorresponding Author: Daniel Patschan, Klinik fur Kardiologie, Angiologie und Nephrologie, Innere Medizin I, Klinikum Brandenburg, Medizinische Hochschule Brandenburg, Hochstrabe 29, Brandenburg 14770, Germany
Manuscript submitted May 6, 2019, accepted August 12, 2019
Short title: Diabetes and Inflammation in AKI
doi: https://doi.org/10.14740/jocmr3852
| Abstract | ▴Top |
Background: Acute kidney injury (AKI) significantly worsens the prognosis of hospitalized patients. Diabetes mellitus (DM) affects a growing number of individuals in the western world. DM subjects are at a higher risk for acquiring AKI during the stay at the hospital. The current study intended to quantify serum levels of specific immunomodulatory cytokines in diabetic mice suffering from AKI.
Methods: DM was induced in male C57/Bl6N mice by systemic injections of beta cell-toxic streptozotocin. Animals underwent bilateral renal ischemia (45 min) 6 weeks later.
Results: Post-ischemic diabetic mice showed significantly differing serum concentrations of the majority of all analytes as compared to untreated controls and non-diabetic (post-ischemic) animals.
Conclusions: Together, our data suggest DM-associated immune activation in AKI. One may suppose that inadequate stimulation of the humoral/cellular immune response potentially contributes to the higher ischemia susceptibility of the organ in DM.
Keywords: Acute kidney injury; Systemic inflammation; Insulin; Diabetes mellitus
| Introduction | ▴Top |
Acute kidney injury (AKI) significantly worsens the prognosis of hospitalized patients worldwide; incidences have been increased over the last 10 years [1]. In general, diabetic patients are at higher-than-average AKI risk, not only in terms of acquiring the syndrome per se but also regarding the long-term morbidity and mortality [2]. Although AKI has traditionally been defined as a “tubular” disorder, numerous investigations published in the last 8 - 10 years established AKI-associated inflammation as a new area in the field [3]. Diabetes, without any doubt, compromises the immune response. Patients are particularly susceptible to bacterial and fungal infections, namely sepsis. The current report presents selected results from a recent study on autophagy activation in circulating proangiogenic cells (PACs) [4]. We evaluated numerous AKI-related outcome parameters, including circulating patterns of immunomodulatory cytokines. Herein, we would like to discuss this particular aspect shortly.
| Materials and Methods | ▴Top |
This section will exclusively describe the analyses performed for acquiring the results as presented.
Animals
The protocol followed the guidelines of the German Institute of Health Guide for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee of the University of Gottingen. For all experiments, we employed male C57/Bl6N mice (8 - 12 weeks old). Mice were bred in the local animal facility of the Gottingen University Hospital. Animals were separately caged with a 12:12-h light-dark cycle and had free access to water and chow throughout the study.
Surgical procedures
Anesthesia was performed with the following solution, applied intraperitoneally (IP): 300 µL of 6 mg/100 g ketamine hydrochloride plus 0.77 mg/100 g xylazine hydrochloride. The abdominal cavity was opened by a 1.5-cm midlaparotomy, followed by removal of the right kidney. The left kidney was exposed, and clamping of the renal pedicle was performed with micro serrefines (Fine Science Tools, Forster City, CA, USA). AKI was induced by renal pedicle clamping for 45 min. The procedure has been published several times in the past [5]. After 45 min, the clamps were released, and a constant volume of PAC-containing endothelial basal medium-2 (EBM-2) media (1 × 106 PACs in 100 µL, either native or after zVAD-/MG132-pretreatment) was injected into the tail vein and thus, into the systemic circulation. The abdominal incision was closed, and animals were sacrificed 48 h later.
Induction of diabetes mellitus (DM)
Insulin-dependent DM was induced by IP injections of streptozotocin (STZ, Sigma Chemical Co., Saint Louis, MO, USA; 50 mg/kg) for 5 consecutive days. Blood glucose testing was performed at day 7 after the last injection in order to verify the diabetic deterioration. Only animals with significant hyperglycemia were included and subsequently caged for further 6 weeks before surgery.
Culture of murine PACs
The procedure of culturing PACs has been described numerous times in the past [4, 5].
PAC preconditioning with zVAD and MG132
Cultured PACs intended for cell injection experiments were detached by trypsinization after the first passage. After washing with phosphate-buffered saline (PBS) once, cells were resuspended in 100 µL of EGM-2 for systemic injection or for further in vitro treatment. For in vitro treatment PACs were incubated with either zVAD (20 µmol/L in EGM-2) (InvivoGen) or with MG132 (10 µmol/L in EGM-2) (InvivoGen) for 60 min at 37 °C. After washing the cells once with EGM-2, they were resuspended in 100 µL of EGM-2 for systemic injection.
Cytokine analysis
Serum samples were analyzed for the following cytokines by using the Bio-Plex Multiplex Immunoassay System (BioRad) according to the manufacturer’s instructions: interleukin 1A (IL-1A), IL-1B, IL-3, IL-5, IL-6, IL-10, IL-12p40, IL-12b70, IL-13, IL-17A, interferon-g (INF-g), keratinocyte cytokine (KC), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1a (MIP-1a), MIP-1b, Rantes, tumor necrosis factor-a (TNF-a), and granulocyte colony-stimulating factor (G-CSF). Figure 1 summarizes the methodical approach.
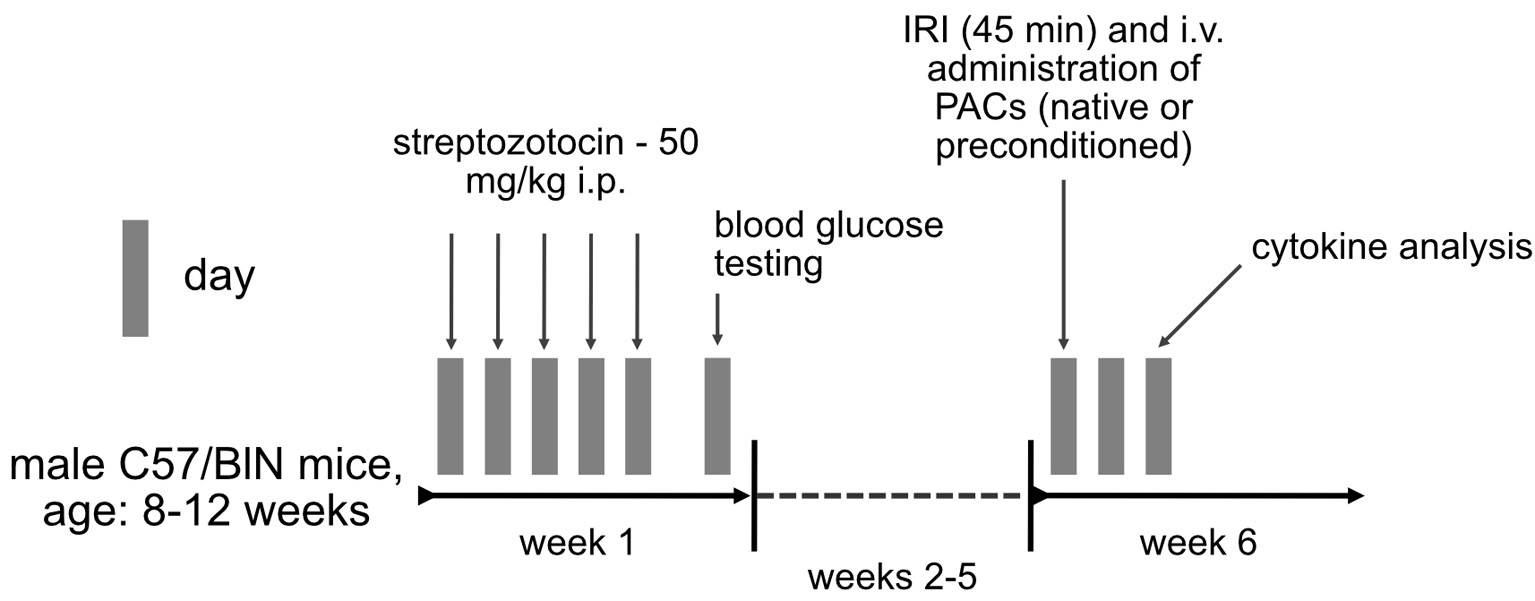 Click for large image | Figure 1. Methodical approach in the current study. Insulin-dependent diabetes mellitus was induced in male C57/Bl6N mice by IP injection of streptozotocin for 5 consecutive days. Blood glucose testing was performed at day 7 after the first injection. Six weeks later, animals underwent bilateral renal ischemia (ischemia reperfusion injury), followed by systemic administration of native or preconditioned syngeneic murine PACs. Cytokine analysis was performed 2 days (48 h) later. IP: introperitoneally; PAC: proangiogenic cell. |
Statistical analysis
All data are given as mean ± the standard error of the mean (SEM). Differences between two groups were quantified by Student’s t-test, and differences between more than two groups were analyzed by analysis of variance (ANOVA). A P value < 0.05 was considered statistically significant.
| Results | ▴Top |
Renal ischemia modulates circulating immunomodulatory cytokines particularly under diabetic circumstances
All, except for three cytokines (IL-17A, KC, Rantes), showed significant inner-group variability. If compared to untreated controls, serum levels of the following cytokines significantly differed in post-ischemic diabetic mice: higher (IL-1A, IL-1B, IL-5, IL-6, IL-10, IL-12p70, IL-13, IFN-g, MCP-1, MIP-1 beta, and G-CSF); lower (IL-12p40). In non-diabetic animals only four cytokines differed from controls post-ischemia in a significant manner: IL-6, MCP-1, MIP-1b, and G-CSF (all higher after ischemia). Finally, the following differences occurred between post-ischemic mice with versus without diabetes: higher (IL-1A, IL-1B, IL-3, IL-6, IL-10, IL-12p70, IL-13, IFN-g, MCP-1, MIP-1 alpha and beta, TNF-a, and G-CSF); lower (IL-12p40). In summary, diabetic circumstances were significantly associated with increased serum concentrations of numerous immunomodulatory cytokines as compared to untreated controls and post-ischemic non-diabetic animals. One cytokine (IL-12p40) was lower in diabetic than in non-diabetic mice.
Serological immunoactivation is not significantly altered by PAC administration
Almost all analytes did not differ between mice without cell therapy and animals injected with native or zVAD/MG132-pretreated syngeneic murine PACs, neither under diabetic nor non-diabetic conditions. The respective data are shown in Figure 2; we will not list all numerical results in detail.
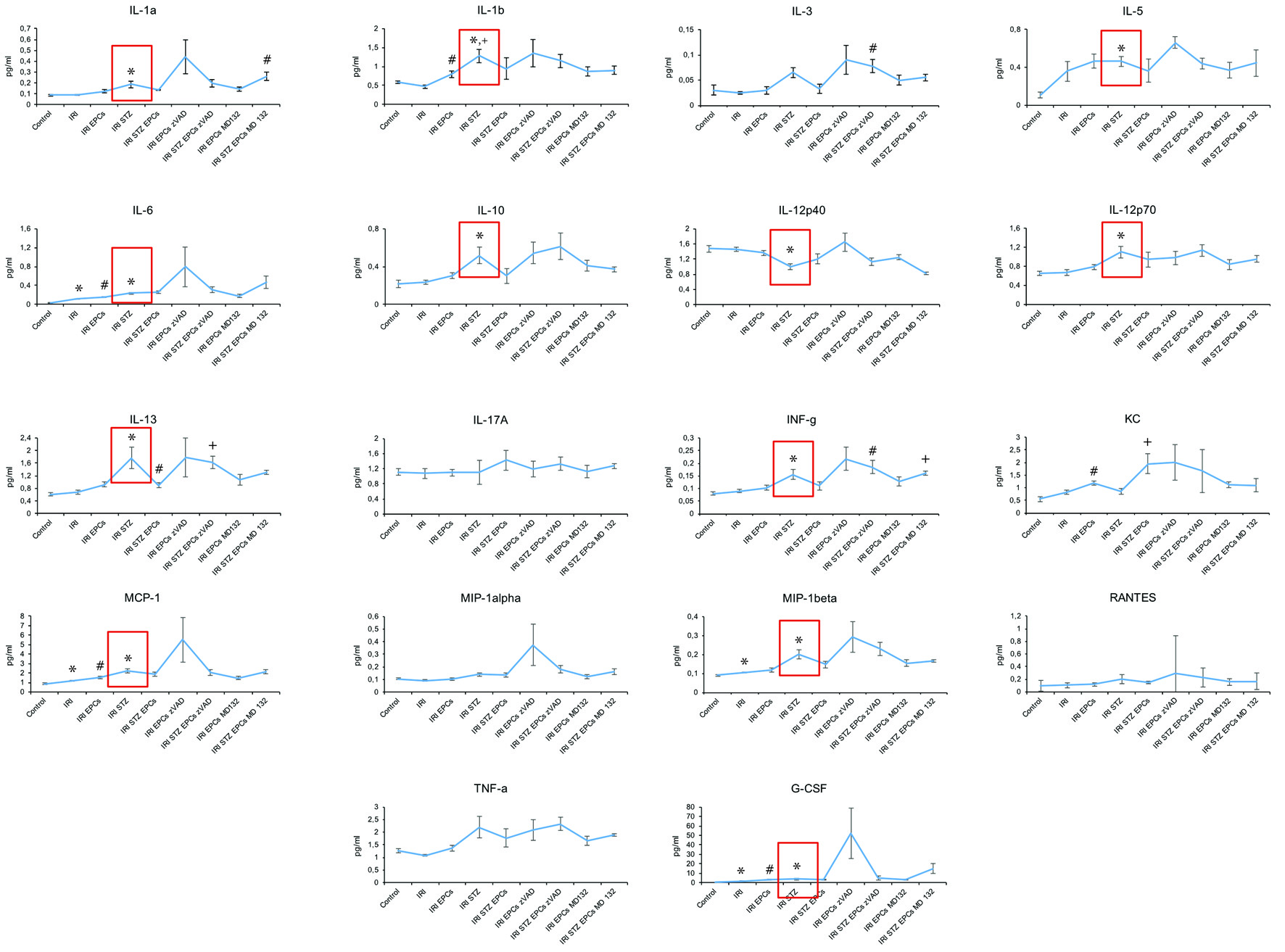 Click for large image | Figure 2. Serum cytokine levels at 48 h after ischemia-reperfusion injury in non-diabetic and diabetic mice. IRI: ischemia reperfusion injury; STZ: streptozotocine; PAC: proangiogenic cell. The symbols indicate significant differences (P < 0.05) in comparison to certain groups: * as compared to “Control”; ° as compared to “IRI”; + as compared to “IRI STZ PACs” in I and K and to “IRI STZ” in L and to “IRI” in R; # as compared to “IRI STZ PACs” in A and to “IRI” in B, E, L, and M (all data as mean ± SEM). |
| Discussion | ▴Top |
The current report intends to provide evidence for significant (serological) immunoactivation in ischemic AKI under diabetic circumstances. The literature on the topic “AKI-associated inflammation in diabetes” is quite limited to put it mildly. Several clinical studies addressed the particular question of whether diabetes increases the AKI risk under either septic or non-septic conditions. In a retrospective, data-based analysis, Mehta et al identified a higher prevalence of DM in AKI subjects versus those without any AKI episode (49% vs. 33%, P < 0.0001) [6]. Oliveira et al [7] also found DM more frequently in AKI patients (19.6% vs. 9.3%, P = 0.007). A 2012 published study by Girman et al retrospectively evaluated yearly AKI incidences in DM versus non DM, and the respective numbers were 192 versus 27/100,000 subjects [8]. In 2015, Venot et al analyzed AKI epidemiology in DM with sepsis/septic shock [9]. Although AKI did not occur more often in diabetics per se, these individuals required dialysis therapy more frequently at the time of discharge. In summary, one may conclude that DM increases the AKI risk in general. For further information, we would like to refer to a review article published in 2016 [2].
From a mechanistic point of view, it seems plausible that diabetics are at higher AKI risk. The syndrome is typically associated with significant macro- and microvascular pathology in the majority of affected individuals. Both atherosclerosis and arteriolosclerosis evolve over years, particularly if blood glucose levels are controlled poorly. Nevertheless, there is experimental evidence suggesting hyperglycemia as “fast-acting” AKI risk factor as well. Shi et al found the renal microvasculature to be critically affected in 10-12-week-old diabetic mice [10]. Thus, the lifespan of these animals was too short for having acquired significant macrovascular damage. The most common cause of AKI, renal hypoperfusion, or ischemia induces numerous functional and structural abnormalities in the kidney. For many years, the “tubulocentric” view dominated the conceptual discussion regarding AKI pathophysiology. In addition to tubular cell dysfunction and damage, renal ischemia typically induces two further pathophysiological events, microvascular dysfunction and interstitial inflammation [11]. Mainly the latter has extensively been studied in the recent past. Virtually all cellular and humoral components of the innate and acquired immune responses are activated in (ischemic) AKI [3]. Most studies on “inflammation in AKI” performed so far have been experimental in nature. However, particular clinical progress became exclusively possible as a direct consequence of immune activation. For instance, (urinary) IL-18 is still evaluated as a new diagnostic marker in AKI subjects [12]. Increased serum IL-6 levels were identified as predictive for septic AKI already 10 years ago [13]. So far, no study systematically analyzed serum levels of specific immunoregulatory cytokines in AKI under diabetic conditions. Our study showed evaluated concentrations of IL-1A, IL-1B, IL-6, IL-10, IL-12p40 and 70, IL-13, IFN-g, MCP-1, MIP-1 beta, and G-CSF in diabetic mice as compared to untreated controls. One single cytokine (IL-12p40) was lower in diabetic animals. Non-diabetic mice differed from controls in exclusively four parameters: IL-6, MCP-1, MIP-1b, and G-CSF, whereas post-ischemic animals with DM significantly differed from post-ischemic non-diabetic mice in a total number of 14 analytes (for details see Results section). The mere amount of parameters does not allow a detailed discussion of any mechanistic implications. However, our data leave no doubt that ischemic AKI is associated with significant immune activation in DM. Hence, future investigations should evaluate the particular role of both the cellular and humoral immune responses in AKI under diabetic circumstances. Regarding the epidemiological situation of AKI in general and of AKI in diabetic subjects specifically, respective studies are mandatory to say the least.
Acknowledgments
None to declare.
Financial Disclosure
The study was supported by the European Foundation for the Study of Diabetes (EFSD).
Conflict of Interest
The authors declare that there is no conflict of interest.
Informed Consent
Not applicable.
Author Contributions
DP designed the study, applied for funding, analyzed data and wrote the manuscript; KS performed all surgical experiments; EH and JCH performed cytokine analyses; SP analyzed data and corrected the manuscript; OR corrected the manuscript and helped with data analysis; GAM corrected the manuscript and supplied laboratory space.
| References | ▴Top |
- Bienholz A, Wilde B, Kribben A. From the nephrologist's point of view: diversity of causes and clinical features of acute kidney injury. Clin Kidney J. 2015;8(4):405-414.
doi pubmed - Patschan D, Muller GA. Acute kidney injury in diabetes mellitus. Int J Nephrol. 2016;2016:6232909.
doi pubmed - Mulay SR, Holderied A, Kumar SV, Anders HJ. Targeting inflammation in so-called acute kidney injury. Semin Nephrol. 2016;36(1):17-30.
doi pubmed - Schwarze K, Kribben A, Ritter O, Muller GA, Patschan D. Autophagy activation in circulating proangiogenic cells aggravates AKI in type I diabetes mellitus. Am J Physiol Renal Physiol. 2018;315(4):F1139-F1148.
doi pubmed - Patschan D, Schwarze K, Henze E, Patschan S, Muller GA. The endothelial-to-mesenchymal transition and endothelial cilia in EPC-mediated postischemic kidney protection. Am J Physiol Renal Physiol. 2016;310(7):F679-F687.
doi pubmed - Mehta RH, Grab JD, O'Brien SM, Bridges CR, Gammie JS, Haan CK, Ferguson TB, et al. Bedside tool for predicting the risk of postoperative dialysis in patients undergoing cardiac surgery. Circulation. 2006;114(21):2208-2216; quiz 2208.
doi pubmed - Oliveira JF, Silva CA, Barbieri CD, Oliveira GM, Zanetta DM, Burdmann EA. Prevalence and risk factors for aminoglycoside nephrotoxicity in intensive care units. Antimicrob Agents Chemother. 2009;53(7):2887-2891.
doi pubmed - Girman CJ, Kou TD, Brodovicz K, Alexander CM, O'Neill EA, Engel S, Williams-Herman DE, et al. Risk of acute renal failure in patients with Type 2 diabetes mellitus. Diabet Med. 2012;29(5):614-621.
doi pubmed - Venot M, Weis L, Clec'h C, Darmon M, Allaouchiche B, Goldgran-Toledano D, Garrouste-Orgeas M, et al. Acute kidney injury in severe sepsis and septic shock in patients with and without diabetes mellitus: a multicenter study. PLoS One. 2015;10(5):e0127411.
doi pubmed - Shi H, Patschan D, Epstein T, Goligorsky MS, Winaver J. Delayed recovery of renal regional blood flow in diabetic mice subjected to acute ischemic kidney injury. Am J Physiol Renal Physiol. 2007;293(5):F1512-1517.
doi pubmed - Patschan D, Kribben A, Muller GA. Postischemic microvasculopathy and endothelial progenitor cell-based therapy in ischemic AKI: update and perspectives. Am J Physiol Renal Physiol. 2016;311(2):F382-394.
doi pubmed - Schrezenmeier EV, Barasch J, Budde K, Westhoff T, Schmidt-Ott KM. Biomarkers in acute kidney injury - pathophysiological basis and clinical performance. Acta Physiol (Oxf). 2017;219(3):554-572.
doi pubmed - Chawla LS, Seneff MG, Nelson DR, Williams M, Levy H, Kimmel PL, Macias WL. Elevated plasma concentrations of IL-6 and elevated APACHE II score predict acute kidney injury in patients with severe sepsis. Clin J Am Soc Nephrol. 2007;2(1):22-30.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.