

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Review
Volume 10, Number 11, November 2018, pages 799-805
Possibility of a New Therapeutic Strategy for Left Ventricular Dysfunction in Type 2 Diabetes
Masaya Sakamotoa, c, Daisuke Matsutania, Yosuke Kayamab
aDivision of Diabetes, Metabolism and Endocrinology, Department of Internal Medicine, Jikei University School of Medicine, 3-25-8 Nishi-Shinbashi, Minato-ku, Tokyo 105-8461, Japan
bDepartment of Cardiology, Jikei University School of Medicine, 3-25-8 Nishi-Shinbashi, Minato-ku, Tokyo 105-8461, Japan
cCorresponding Author: Masaya Sakamoto, Division of Diabetes, Metabolism and Endocrinology, Department of Internal Medicine, Jikei University School of Medicine, 3-25-8 Nishi-Shinbashi, Minato-ku, Tokyo 105-8461, Japan
Manuscript submitted August 16, 2018, accepted September 7, 2018
Short title: Left Ventricular Dysfunction in Diabetes
doi: https://doi.org/10.14740/jocmr3584w
| Abstract | ▴Top |
Type 2 diabetes mellitus (T2DM) substantially increases the risk of cardiovascular events, including heart failure (HF), due to complications such as hypertension, obesity and dyslipidemia based on metabolic syndrome, which plays the central pathological role in HF. A reason is that T2DM causes left ventricular (LV) diastolic dysfunction beginning in the early phase of the disease, which in turn increases the risk of development of HF independently of the control of blood glucose levels, blood pressure or the presence of coronary artery diseases. Intracellular metabolic disorders and increased oxidative stress due to hyperglycemia, increased insulin resistance and chronic inflammation are pathogenic mechanisms involved in the LV diastolic dysfunction caused by T2DM. These mechanisms lead to structural changes in the heart such as LV hypertrophy and interstitial fibrosis, resulting in HF. The prevalence of HF with preserved ejection fraction (HFpEF), the major pathology of LV diastolic dysfunction, has been increasing recently, and a high incidence of HFpEF in patients with T2DM was reported. An effective therapy has not been established for HFpEF because multiple comorbidities such as advanced age, hypertension, obesity, dyslipidemia, chronic kidney disease and atrial fibrillation as well as diabetes are involved in its pathology. In the present review, we review the involvement of associated conditions such as hypertension, obesity and advanced age from the aspect of the T2DM and LV diastolic dysfunction and discuss the possibility of the development of a new therapeutic strategy for LV diastolic dysfunction and HFpEF.
Keywords: Left ventricular diastolic dysfunction; Type 2 diabetes mellitus; Heart failure with preserved ejection fraction; Sodium-glucose cotransporter 2 inhibitors; Imeglimin
| Introduction | ▴Top |
The prevalence of heart failure (HF) with preserved ejection fraction (HFpEF), the major pathophysiology of left ventricular (LV) diastolic dysfunction, has been increasing recently, and a high incidence of HFpEF in patients with type 2 diabetes mellitus (T2DM) was reported [1, 2]. Although the risk factor of patients with HFpEF and HF with reduced ejection fraction (HFrEF) are similar to some extent, such as hypertension and LV hypertrophy [3, 4]. On the other hand, it was also pointed out that the backgrounds of patients with HFrEF and HFpEF basically differed in the age of onset, sex and causes of death, and also that the pathology fundamentally differs between these types of HF [5, 6]. HFrEF is a contractile dysfunction with cardiac diseases such as ischemic heart disease, dilated cardiomyopathy and valvular heart disease as major causes. Various systemic conditions such as diabetes, advanced age, hypertension, obesity, dyslipidemia and chronic kidney disease are involved in the onset of HFpEF. These induce chronic inflammation and increased oxidative stress in the heart and blood vessels and secondarily lead to vascular endothelial damage, LV hypertrophy and interstitial fibrosis. It was speculated that these events finally result in the onset of HFpEF [7], but the detailed mechanism is still unknown. As various extra-cardiac factors are involved in the onset of HFpEF, large-scale clinical trials failed to show reductions in mortality in patients with HFpEF by angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), β-blockers or aldosterone antagonists, which are cardioprotective drugs that have shown effectiveness in treating patients with HFrEF [8-10]. Meanwhile, there have been large-scale clinical trials of sodium glucose transporter 2 (SGLT2) inhibitors that not only exert a blood glucose lowering effect but also an improvement in systemic metabolism that did show a favorable effect. The Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patient-Removing Excess Glucose (EMPA-REG OUTCOME) study [11, 12] and the Canagliflozin Cardiovascular Assessment Study (CANVAS) [13] reported that these drugs suppressed the onset of cardiovascular (CV) events (especially hospitalization due to HF) in a short period of time in patients with T2DM who were at high risk for a CV event and were being treated for its secondary prevention. Currently, the findings that SGLT2 inhibitors suppressed the onset of HF in patients with T2DM are being analyzed from various viewpoints, and expectation is running high for the preventive effect of SGLT2 inhibitors on the onset of HFpEF. Supporting this expectation, a recent study demonstrated that a SGLT2 inhibitor improved LV diastolic function and LV hypertrophy in patients with T2DM [14, 15]. In the present review, we review the involvement of associated conditions such as hypertension, obesity and advanced age from the aspect of the T2DM and LV diastolic dysfunction and discuss the possibility of the development of a new therapeutic strategy for LV diastolic dysfunction and HFpEF.
| Factors related to left ventricular diastolic dysfunction | ▴Top |
Diabetes
The Framingham Heart Study revealed that the risk for HF was increased two- and five-fold among male and female patients with T2DM, respectively. Interestingly, more patients with diabetes were noted to develop chronic HF within 30 months of observation period [16], indicating a close relationship between diabetes and HF [17, 18]. Mortality was shown to be high in patients with T2DM whose E/e’ ratio, an index of diastolic function in an echocardiographic examination, was larger than 15. Furthermore, since the influence of diabetes on hospitalizations due to HF or CV death is greater in patients with HFpEF than in those with HFrEF, diabetes may be a critical factor in the development of LV dysfunction and its progression to HFpEF. The relationship between diabetes and HF is thought to be multifactorial so that numerous mechanisms have been reported to play an important role in its etiology. Diabetes leads to cardiac structural and functional disturbances in the myocardium. It is widely recognized that diabetic cardiomyopathy consists of cardiomyocyte hypertrophy, inflammation, fibrosis, increased apoptosis and metabolic abnormalities [19-21]. Cardiac inflammation induced by pro-inflammatory cytokines and chemokines may contribute to the pathogenesis of pressure-overload induced HF as well as diabetic cardiomyopathy [22-24]. Indeed, concentrations of inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-alpha) and interleukin (IL)-6, are increased in the serum of diabetic patients, suggesting a link between systemic inflammation and LV diastolic dysfunction [25]. LV hypertrophy (LVH) is a characteristic feature of the morphologic manifestations of diabetic cardiomyopathy, generally representing a more advanced stage of disease. Echocardiographic evidence revealed that LVH is a common structural and functional alteration in diabetic patients even in the absence of coronary artery disease or hypertension [26]. Although LVH is frequently associated with increased afterload in diabetic patients with hypertension [27], it can also occur independently of pressure overload [26]. In the diabetic state, neurohormonal activation is seen both at the systemic and tissue levels, and includes up-regulation of the renin-angiotensin-aldosterone system (RAAS), endothelin-1 (ET-1) and the sympathetic nervous system [28-30]. Enhanced activity of local RAAS induces functional abnormalities in the diabetic heart. High glucose concentrations can increase RAAS activation and production of angiotensin-II (AII) in cardiomyocytes and cardiac fibroblasts [31]. Collectively, they contribute to the functional impairments associated with LV dysfunction in diabetic cardiomyopathy.
Obesity
Epidemiological studies have reported that the risk of HF was increased two-fold by obesity and insulin resistance resulting from obesity compared with normal healthy individuals; also, the risk of HF was further increased with elevations in the body mass index [32, 33]. It was reported that insulin resistance was induced even in patients with HF who did not have concomitant diabetes and that insulin resistance makes patients prone to the development of diabetes [34]. Therefore, there may be a close relationship between insulin resistance induced by obesity or diabetes and HF. The pathogenic mechanisms of LV dysfunction and the development of HF are complex (Fig. 1). The secretion of inflammatory cytokines such as IL-6 is enhanced due to obesity based on metabolic syndrome, and systemic insulin resistance is initially elevated. Then, this insulin resistance further promotes insulin secretion causing an insulin-excess state (Fig. 1). The systemic insulin-excess state induces abnormalities in intracellular insulin signal transduction in various tissues. Similarly, it induces increased fatty acid oxidation, decreased glucose uptake and intracellular metabolic impairment in the myocardium. Accordingly, there are then increases in reactive oxidative stress and the induction of mitochondrial dysfunction that results in reduced ATP production and LV dysfunction [35, 36]. In addition, it was reported that cardiomyocytic insulin signal transduction was activated in patients with HF, and that excess insulin signaling contributed to the worsening of LV function [37]. Meanwhile, the activation of the sympathetic nervous system, enhancement of the RAAS, and the associated retention of sodium and increased circulating plasma volume occur following enhanced secretion of inflammatory cytokines and aggravation of insulin resistance. These changes induce LV hypertrophy and interstitial fibrosis characterized by deposition of collagen in interstitial tissues (Fig. 1). Since aerobic metabolism is also impaired in hypertrophied cardiac muscles induced by obesity [33], a decrease in LV active relaxation and an increase in LV stiffness take place, resulting in the appearance of LV diastolic dysfunction. It was reported that morphological observation by echocardiography revealed findings of abnormalities in diastolic function (reduced E/A ratio, prolonged E wave deceleration time, reduced E’ and increased E/e’, and prolonged isovolumic relaxation time (IRT) in obese individuals though echocardiology failed to demonstrate a reduced LVEF). These findings also revealed that diastolic dysfunction became more severe with elongation of the duration of the obese period [38]. As for the morphology of LV hypertrophy due to obesity, the concentric type hypertrophy is more prevalent than the eccentric type. This may be caused by chronic left atrial overload due to increased circulating blood volume caused by obesity and LV diastolic dysfunction.
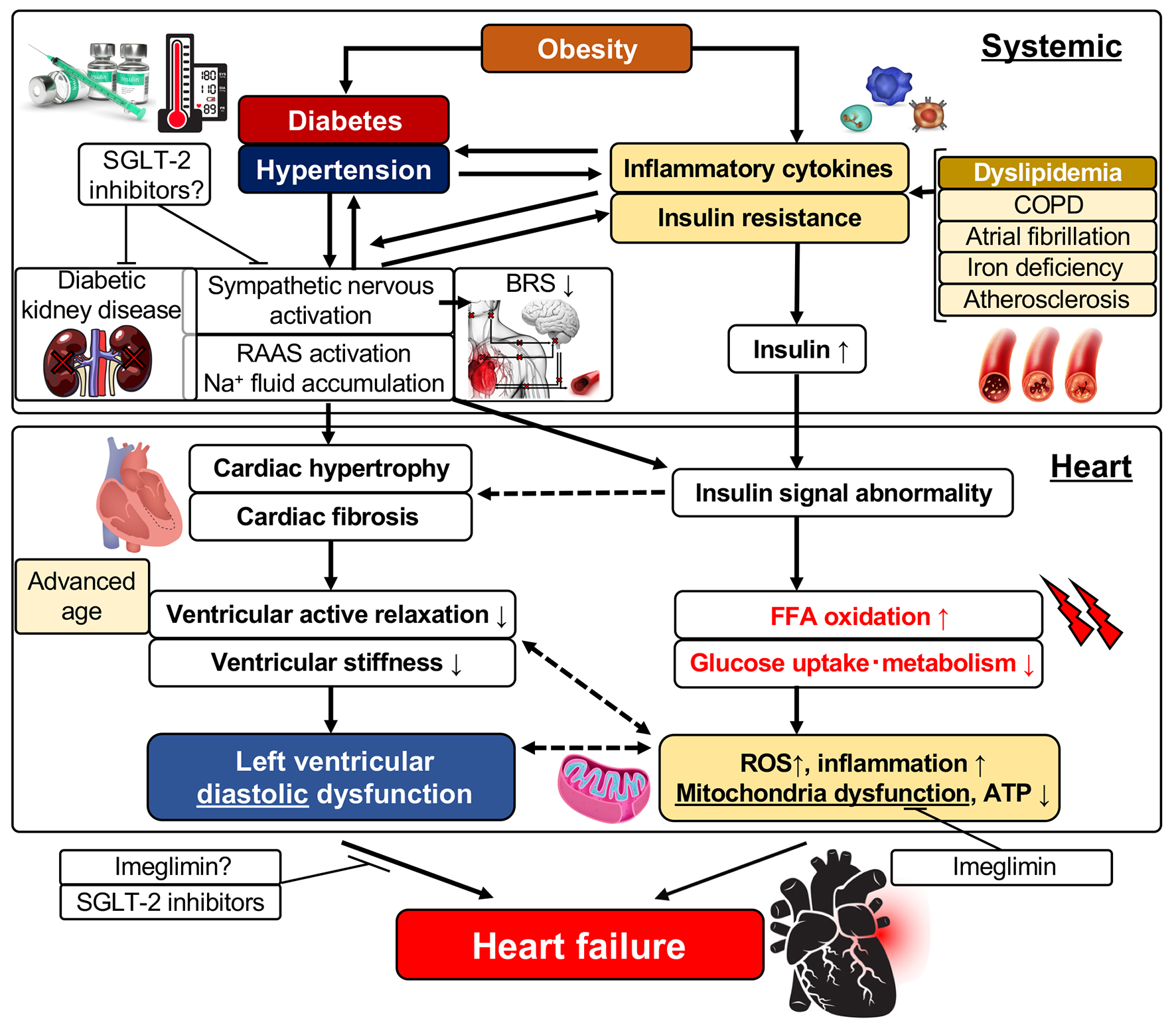 Click for large image | Figure 1. Left ventricular diastolic dysfunction and its related factors. FFA: free fatty acid; ROS: reactive oxygen species; ATP: adenosine triphosphate; SGLT: sodium-glucose cotransporter; RAAS: renin-angiotensin-aldosterone system; IL: interleukin; BRS: baroreflex sensitivity; COPD: chronic obstructive pulmonary disease. |
Hypertension
The relationship between HF and hypertension has long been known. It has been reported that LV hypertrophy associated with hypertension is potentiated by obesity and/or diabetes, and their coexistence was reported to worsen LV diastolic function [39]. Two major mechanisms have been postulated for LV dysfunction caused by hypertension: a mechanism in which pressure load due to hypertension induces LV hypertrophy and HF and a second mechanism whereby increased oxidative stress following RAAS activation and increased secretion of inflammatory cytokines induce LV dysfunction.
Angiogenesis in the myocardium plays a significant role in the mechanism of the development of LV hypertrophy and HF following pressure load [40]. The myocardium exerts a compensatory hypertrophic response to pressure overload, and it also regulates myocardial angiogenesis during the hypertrophic process. Hypoxia-induced factor-1 (HIF-1) induced by hypoxic stimulus and vascular endothelial growth factor (EGF) play important roles in intra-myocardial angiogenesis. Angiogenesis advances with progression of LV hypertrophy. With further progression of LV hypertrophy, however, p53, a tumor suppressor gene, is activated, the expression of HIF-1 located downstream of p53 is suppressed and the expression of VEGF is lowered. It has been thought that HF develops with the reduction of angiogenesis in the heart through these processes. Moreover, various stimuli are known to induce myocardial hypertrophy, myocardial fibrosi, and apoptosis including neurohormonal factors and cytokines, such as AII, ET-1, TNF-alpha activation of redox-sensitive protein kinases and mechanical stretch [41, 42]. Reactive oxygen species (ROS)-induced activation of apoptosis signal-regulating kinase 1 (ASK1) may promote troponin T phosphorylation and be implicated in contractile dysfunction. The AII-associated ROS pathway may influence alterations in the structure and function of excitation-contraction coupling and ionic homeostasis and Ca handling [43].
Advanced age
Many studies have examined changes in cardiac structure and contractile/diastolic function of the heart with aging [44]. During the process of adulthood to senescence, the weight of the myocardium increases due to elevation of systemic blood pressure caused by increases in peripheral vascular resistance. At the same time, the myocardial mass/volume ratio increases due to the decrease in the LV end-diastolic diameter [45]. Regarding the LV contractile function, age-related changes in the EF or fractional shortening was reported, but the influence of aging is small on contractility indices on echocardiography. These indices are thought to be constant throughout the life span [46], whereas LV diastolic function has been known to be more strongly affected by cardiovascular mechanical changes due to aging in comparison with contractile functions. Indices of echocardiography reflecting LV diastolic function are as follows: E wave (early diastolic trans-mitral flow velocity) obtained from mitral annular velocity; A wave; E/A ratio; e’ velocity (mitral annular velocity) obtained by a tissue Doppler method; E/e’ ratio; and the pulmonary venous flow pattern. In adulthood, height of the E wave and A wave decreases and increases, respectively, and the E/A ratio gradually decreases with aging. The LV deceleration time (LV-DT) is prolonged [47]. The E/A ratio continues to increase until about 20 years after birth because the height of the E wave and A wave increases and decreases, respectively. Since the mitral annular velocity, e’, which reflects movement of the entire left ventricle, is less influenced by preload compared with the E/A ratio, it has been considered a better index to assess LV relaxation, but e’ decreases like the E/A ratio and E/e’ ratio increases with aging [48].
Aortic stiffness
HFpEF is HF that develops mainly by diastolic dysfunction in the left ventricle. The characteristic changes in the arterial system brought about by factors such as hypertension and arteriosclerosis are important as causes of diastolic dysfunction. Thus, HFpEF is thought by some to be a vascular disease. HFpEF often develops based on hypertension, and it more frequently develops in elderly persons and females. The reason for the frequent occurrence of HFpEF in elderly persons can be explained by the pulse wave propagation theory. The ascending aorta expands by ejection of blood from the heart to the ascending aorta, and the expansion propagates through the aorta to its periphery as a pulse wave. The speed of propagation that is defined as pulse wave velocity (PWV) depends on stiffness of the aorta. Since the vascular wall is harder in elderly persons than in young adults, the PWV is higher in the former than the latter. Normally, a reflected pulse wave returns to the aorta in the diastolic phase, but it returns in the late systolic phase in elderly persons because of their high PWV. This phenomenon amplifies pulse pressure in the late systolic phase and increases the LV afterload, leading to the development of diastolic HF. Reasons for the frequent development of HFpEF in females are their shorter stature and shorter length of the aorta than in males. Therefore, a reflected pulse wave easily overlaps on the late systolic phase.
In humans, aortic stiffness and pulse pressure increases with aging. Aortic stiffness can be expressed by effective end-systolic elastance (Ea). Thus, Ea reflects both the average and the pulse pressure component of the afterload. Ejection of blood into a stiffened arterial system induces LV remodeling. End-systolic pressure is more highly elevated in elderly persons than in young adults [49]. This excessive elevation of blood pressure is the reason for worsening of LV relaxation and the elevation of end-diastolic LV pressure [50]. This is indubitably one of the mechanisms for the development of HF in elderly persons, that is, resulting in HFpEF (Fig. 1).
Future strategies for treatment of HFpEF
As mentioned above, multiple comorbidities such as advanced age, obesity, hypertension and atherosclerosis as well as diabetes are involved in the development of HFpEF. LV hypertrophy and cardiac interstitial fibrosis have been thought to be the main pathogenesis of HFpEF from the pathological viewpoint. Therefore, inhibitors of the RAAS (ACE inhibitors, ARBs and aldosterone antagonists) have been expected to be effective therapeutic agents for HFpEF because their effectiveness in suppressing LV hypertrophy and interstitial fibrosis was already proven [51, 52]. The results of trials that examined the effectiveness of these agents were published as PEP-CHF [8], CHARM-Preserved [9], I-Preserved [53] and TOPCAT [10], but no trial could demonstrate an improvement in long-term outcome of patients with HFpEF through the use of these drugs. These results have forced us to change the therapeutic strategy for HFpEF. It is necessary to introduce systemic therapeutic interventions or interventions targeted to tissues other than the myocardium as well as using conventional treatments focusing on the regression of LV hypertrophy and LV diastolic dysfunction. It was reported that patients with HFpEF presented with LV stiffening in the systolic phase and had arterial stiffening causing an excessive response of blood pressure to exercise and shift of the diastolic ventricular pressure-volume loop upward [50]. Therefore, it appeared reasonable to focus on the treatment of vascular systems or atherosclerosis as therapy for HFpEF rather than focus on the LV myocardium itself based on a previous report [54]. Noteworthy is a recent study on the treatment of HF and CV diseases targeting adipose tissue. Inflammation dependent on p53, a tumor suppressor gene, occurs in adipose tissues in patients with HF, and the persistent inflammation induces insulin resistance and further aggravates HF [55]. In addition, recent attention has been paid to the relationship between CV diseases and brown adipose tissue. Brown adipose tissue is involved in the elimination of lipids and carbohydrate from circulating blood, and may be an important endothelial organ that ordinarily acts to maintain systemic homeostasis [56]. Since it was reported that brown adipose tissue showed whitening and dysfunction in the state of obesity or HF [57], the improvement of inflammation and metabolism in fat tissues may be an important new therapeutic target for obesity- and diabetes-induced HF.
Large-scale clinical trials using SGLT inhibitors, the EMPA-REG trial and the CANVAS trial, taught us that therapeutic interventions aiming at the improvement of systemic metabolism, including the blood glucose level, blood pressure, lipid levels and insulin resistance, are possibly effective for preventing and improving LV diastolic dysfunction and HFpEF. In our recent trial that examined the effect of a SGLT-2 inhibitor on LV diastolic function and sympathetic nerve activity in patients with T2DM, the inhibitor significantly prevented worsening of LV diastolic function without worsening of sympathetic nerve activity such as that indicated by the heart rate and baroreflex sensitivity in T2DM patients at high risk for CV events [58]. Furthermore, the administration of the SGLT-2 inhibitor significantly improved LV diastolic function in the subgroup of patients whose hemoglobin level increased, indicating that levels of erythropoietic hormones, including erythropoietin, and sympathetic nerve activity are related to these results. Imeglimin is a novel drug that belongs to a new drug class called glimins authorized by the World Health Organization. Imeglimin, which is now under clinical trials, has the unique mechanism of improving mitochondrial functions and acts to potentiate blood glucose concentration-dependent insulin secretion, to improve insulin resistance, and to suppress gluconeogenesis in three major organs (liver, muscle and pancreas) that play important roles in the treatment of T2DM. Imeglimin is thereby expected to exert a blood glucose lowering effect. Its effect of improving vascular endothelial function was already reported as one of its versatile effects other than lowering blood glucose values [59]. It was shown that mitochondrial function is lower in the myocardium with LV diastolic dysfunction, which is the characteristic feature of diabetic cardiomyopathy [60]. In addition, it was recently reported that mitochondrial oxidative stress induced insulin resistance and LV diastolic dysfunction [61], and that mitochondrial oxidative stress was also involved in diastolic dysfunction and cardiac mitophagy [62]. Therefore, imeglimin is expected to be a potential therapeutic agent for HFpEF to prevent LV dysfunction in its early phase (Fig. 1).
Conflict of Interest
The authors of this manuscript have the following competing interests: MS has participated in speaker’s bureaus/advisory panels for Sanofi, Daiichi-Sankyo, Astellas and Tanabe-Mitsubishi.
| References | ▴Top |
- Bertoni AG, Hundley WG, Massing MW, Bonds DE, Burke GL, Goff DC, Jr. Heart failure prevalence, incidence, and mortality in the elderly with diabetes. Diabetes Care. 2004;27(3):699-703.
doi pubmed - Dhingra A, Garg A, Kaur S, Chopra S, Batra JS, Pandey A, Chaanine AH, et al. Epidemiology of heart failure with preserved ejection fraction. Curr Heart Fail Rep. 2014;11(4):354-365.
doi pubmed - Shah AM, Solomon SD. Phenotypic and pathophysiological heterogeneity in heart failure with preserved ejection fraction. Eur Heart J. 2012;33(14):1716-1717.
doi pubmed - Shah AM, Pfeffer MA. The many faces of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2012;9(10):555-556.
doi pubmed - Juilliere Y, Venner C, Filippetti L, Popovic B, Huttin O, Selton-Suty C. Heart failure with preserved ejection fraction: A systemic disease linked to multiple comorbidities, targeting new therapeutic options. Arch Cardiovasc Dis. 2018.
doi - Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, Metra M, et al. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol. 2018;72(10):1081-1090.
doi pubmed - Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263-271.
doi pubmed - Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J, Investigators P-C. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. 2006;27(19):2338-2345.
doi pubmed - Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet. 2003;362(9386):777-781.
doi - Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell N, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370(15):1383-1392.
doi pubmed - Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117-2128.
doi pubmed - Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, Johansen OE, et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME(R) trial. Eur Heart J. 2016;37(19):1526-1534.
doi pubmed - Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(7):644-657.
doi pubmed - Matsutani D, Sakamoto M, Kayama Y, Takeda N, Horiuchi R, Utsunomiya K. Effect of canagliflozin on left ventricular diastolic function in patients with type 2 diabetes. Cardiovasc Diabetol. 2018;17(1):73.
doi pubmed - Verma S, Garg A, Yan AT, Gupta AK, Al-Omran M, Sabongui A, Teoh H, et al. Effect of empagliflozin on left ventricular mass and diastolic function in individuals with diabetes: an important clue to the EMPA-REG OUTCOME trial? Diabetes Care. 2016;39(12):e212-e213.
doi pubmed - Nichols GA, Hillier TA, Erbey JR, Brown JB. Congestive heart failure in type 2 diabetes: prevalence, incidence, and risk factors. Diabetes Care. 2001;24(9):1614-1619.
doi pubmed - Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34(1):29-34.
doi - Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. 1979;241(19):2035-2038.
doi - Suzuki H, Kayama Y, Sakamoto M, Iuchi H, Shimizu I, Yoshino T, Katoh D, et al. Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes. 2015;64(2):618-630.
doi pubmed - Rajesh M, Mukhopadhyay P, Batkai S, Patel V, Saito K, Matsumoto S, Kashiwaya Y, et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol. 2010;56(25):2115-2125.
doi pubmed - McQueen AP, Zhang D, Hu P, Swenson L, Yang Y, Zaha VG, Hoffman JL, et al. Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J Mol Cell Cardiol. 2005;39(6):882-892.
doi pubmed - Kayama Y, Minamino T, Toko H, Sakamoto M, Shimizu I, Takahashi H, Okada S, et al. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J Exp Med. 2009;206(7):1565-1574.
doi pubmed - Haffner SM. The metabolic syndrome: inflammation, diabetes mellitus, and cardiovascular disease. Am J Cardiol. 2006;97(2A):3A-11A.
doi pubmed - Khullar M, Al-Shudiefat AA, Ludke A, Binepal G, Singal PK. Oxidative stress: a key contributor to diabetic cardiomyopathy. Can J Physiol Pharmacol. 2010;88(3):233-240.
doi pubmed - Dinh W, Futh R, Nickl W, Krahn T, Ellinghaus P, Scheffold T, Bansemir L, et al. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol. 2009;8:58.
doi pubmed - Eguchi K, Boden-Albala B, Jin Z, Rundek T, Sacco RL, Homma S, Di Tullio MR. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. Am J Cardiol. 2008;101(12):1787-1791.
doi pubmed - Bell DS. Hypertension and diabetes: a toxic combination. Endocr Pract. 2008;14(8):1031-1039.
doi pubmed - Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93(1):137-188.
doi pubmed - Wang J, Song Y, Wang Q, Kralik PM, Epstein PN. Causes and characteristics of diabetic cardiomyopathy. Rev Diabet Stud. 2006;3(3):108-117.
doi pubmed - van der Horst IC, de Boer RA, Hillege HL, Boomsma F, Voors AA, van Veldhuisen DJ. Neurohormonal profile of patients with heart failure and diabetes. Neth Heart J. 2010;18(4):190-196.
doi pubmed - Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57(12):3297-3306.
doi pubmed - Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, et al. Obesity and the risk of heart failure. N Engl J Med. 2002;347(5):305-313.
doi pubmed - Ingelsson E, Sundstrom J, Arnlov J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA. 2005;294(3):334-341.
doi pubmed - Witteles RM, Fowler MB. Insulin-resistant cardiomyopathy clinical evidence, mechanisms, and treatment options. J Am Coll Cardiol. 2008;51(2):93-102.
doi pubmed - Young ME, McNulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation. 2002;105(15):1861-1870.
doi pubmed - Sharma N, Okere IC, Duda MK, Chess DJ, O'Shea KM, Stanley WC. Potential impact of carbohydrate and fat intake on pathological left ventricular hypertrophy. Cardiovasc Res. 2007;73(2):257-268.
doi pubmed - Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, Tateno K, et al. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest. 2010;120(5):1506-1514.
doi pubmed - Orhan AL, Uslu N, Dayi SU, Nurkalem Z, Uzun F, Erer HB, Hasdemir H, et al. Effects of isolated obesity on left and right ventricular function: a tissue Doppler and strain rate imaging study. Echocardiography. 2010;27(3):236-243.
doi pubmed - Liu JE, Palmieri V, Roman MJ, Bella JN, Fabsitz R, Howard BV, Welty TK, et al. The impact of diabetes on left ventricular filling pattern in normotensive and hypertensive adults: the Strong Heart Study. J Am Coll Cardiol. 2001;37(7):1943-1949.
doi - Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446(7134):444-448.
doi pubmed - Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation. 1997;95(5):1247-1252.
doi pubmed - Zhang W, Elimban V, Nijjar MS, Gupta SK, Dhalla NS. Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp Clin Cardiol. 2003;8(4):173-183.
pubmed - Kayama Y, Raaz U, Jagger A, Adam M, Schellinger IN, Sakamoto M, Suzuki H, et al. Diabetic cardiovascular disease induced by oxidative stress. Int J Mol Sci. 2015;16(10):25234-25263.
doi pubmed - Okura H, Takada Y, Yamabe A, Kubo T, Asawa K, Ozaki T, Yamagishi H, et al. Age- and gender-specific changes in the left ventricular relaxation: a Doppler echocardiographic study in healthy individuals. Circ Cardiovasc Imaging. 2009;2(1):41-46.
doi pubmed - Daimon M, Watanabe H, Abe Y, Hirata K, Hozumi T, Ishii K, Ito H, et al. Normal values of echocardiographic parameters in relation to age in a healthy Japanese population: the JAMP study. Circ J. 2008;72(11):1859-1866.
doi pubmed - Ruan Q, Nagueh SF. Effect of age on left ventricular systolic function in humans: a study of systolic isovolumic acceleration rate. Exp Physiol. 2005;90(4):527-534.
doi pubmed - Mantero A, Gentile F, Gualtierotti C, Azzollini M, Barbier P, Beretta L, Casazza F, et al. Left ventricular diastolic parameters in 288 normal subjects from 20 to 80 years old. Eur Heart J. 1995;16(1):94-105.
doi pubmed - Kuznetsova T, Herbots L, Lopez B, Jin Y, Richart T, Thijs L, Gonzalez A, et al. Prevalence of left ventricular diastolic dysfunction in a general population. Circ Heart Fail. 2009;2(2):105-112.
doi pubmed - Chen CH, Nakayama M, Nevo E, Fetics BJ, Maughan WL, Kass DA. Coupled systolic-ventricular and vascular stiffening with age: implications for pressure regulation and cardiac reserve in the elderly. J Am Coll Cardiol. 1998;32(5):1221-1227.
doi - Kawaguchi M, Hay I, Fetics B, Kass DA. Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction: implications for systolic and diastolic reserve limitations. Circulation. 2003;107(5):714-720.
doi pubmed - Hogg K, McMurray J. Neurohumoral pathways in heart failure with preserved systolic function. Prog Cardiovasc Dis. 2005;47(6):357-366.
doi pubmed - Little WC, Brucks S. Therapy for diastolic heart failure. Prog Cardiovasc Dis. 2005;47(6):380-388.
doi pubmed - Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359(23):2456-2467.
doi pubmed - Burkhoff D, Maurer MS, Packer M. Heart failure with a normal ejection fraction: is it really a disorder of diastolic function? Circulation. 2003;107(5):656-658.
doi pubmed - Shimizu I, Yoshida Y, Katsuno T, Tateno K, Okada S, Moriya J, Yokoyama M, et al. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab. 2012;15(1):51-64.
doi pubmed - Shimizu I, Walsh K. The whitening of brown fat and its implications for weight management in obesity. Curr Obes Rep. 2015;4(2):224-229.
doi pubmed - Shimizu I, Aprahamian T, Kikuchi R, Shimizu A, Papanicolaou KN, MacLauchlan S, Maruyama S, et al. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest. 2014;124(5):2099-2112.
doi pubmed - Matsutani D, Sakamoto M, Minato S, Kayama Y, Takeda N, Horiuchi R, Utsunomiya K. Visit-to-visit HbA1c variability is inversely related to baroreflex sensitivity independently of HbA1c value in type 2 diabetes. Cardiovasc Diabetol. 2018;17(1):100.
doi pubmed - Detaille D, Vial G, Borel AL, Cottet-Rouselle C, Hallakou-Bozec S, Bolze S, Fouqueray P, et al. Imeglimin prevents human endothelial cell death by inhibiting mitochondrial permeability transition without inhibiting mitochondrial respiration. Cell Death Discov. 2016;2:15072.
doi pubmed - Flarsheim CE, Grupp IL, Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Physiol. 1996;271(1 Pt 2):H192-202.
pubmed - Kato ET, Giugliano RP, Ruff CT, Koretsune Y, Yamashita T, Kiss RG, Nordio F, et al. Efficacy and safety of edoxaban in elderly patients with atrial fibrillation in the ENGAGE AF-TIMI 48 trial. J Am Heart Assoc. 2016;5(5):e003432.
doi - Koncsos G, Varga ZV, Baranyai T, Boengler K, Rohrbach S, Li L, Schluter KD, et al. Diastolic dysfunction in prediabetic male rats: Role of mitochondrial oxidative stress. Am J Physiol Heart Circ Physiol. 2016;311(4):H927-H943.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.