

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Case Report
Volume 10, Number 11, November 2018, pages 848-852
Fashionably Late: A Case of Delayed Cutaneous Manifestations in Juvenile Dermatomyositis
Maya Antoinea, Patrick T Reevesa, f, Luis Rohenaa, b, c, Olcay Jonesd, Brian Fauxa, c, e
aDepartment of Pediatrics, San Antonio Uniformed Services Health Education Consortium, San Antonio, TX, USA
bDivision of Genetics, San Antonio Uniformed Services Health Education Consortium, San Antonio, TX, USA
cDepartment of Pediatrics, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA
dDivision of Pediatric Rheumatology, Walter Reed National Military Medical Center, Bethesda, MA, USA
eDivision of Child Neurology, San Antonio Uniformed Services Health Education Consortium, San Antonio, TX, USA
fCorresponding Author: Patrick T Reeves, 3551 Roger Brooke Drive, JBSA Ft Sam Houston, TX 78234, USA
Manuscript submitted July 26, 2018, accepted August 23, 2018
Short title: Delayed Manifestations in Juvenile Dermatomyositis
doi: https://doi.org/10.14740/jocmr3547w
| Abstract | ▴Top |
Juvenile dermatomyositis (JDM) is a rare, but well recognized multi-systemic inflammatory myopathy in children defined by proximal muscle weakness and distinctive skin lesions, that if recognized and treated early result in decreased morbidity and mortality. The 1975 criteria established by Bohan and Peter center around the propensity for early development of heliotrope and Gottron’s lesions in combination with specific laboratory abnormalities, and are still the leading diagnostic tool. The following case demonstrates a toddler with an atypical presentation of JDM in which delayed dermatologic manifestations hindered initial diagnosis. A previously healthy 2 years and 11 months old female presented to the emergency department with a 7-month history of bilateral knee pain and progressive muscular weakness. Initial evaluation yielded a diagnosis of idiopathic rhabdomyolysis but progressive deterioration prompted additional workup. During her course of care, the patient required admission at numerous facilities for specialty procedures including swallow studies, electromyography, Nissen fundoplication with G-tube insertion, and eventual muscle biopsy, resulting in pathology clinching the diagnosis. Post-diagnosis the development of a heliotrope and malar rash ensued, 11 months after commencement of original presentation. As the Bohan and Peter criteria of 1975 can help to aid in diagnosis of JDM for textbook presentations, atypical cases such as ours suggest that revision to current diagnostic criteria needs to be established. Also, with many pediatric rheumatologists opting for less invasive methods than muscle biopsy to aid in diagnosis, in combination with the heterogeneous nature of currently tracked serous markers, the risk for delayed or missed diagnosis is amplified. As prior research has demonstrated, early diagnosis leads to better outcomes for children battling JDM. Therefore, it is vital that criteria be revised and additional research be conducted for more sensitive and specific markers to help aid in early diagnosis of JDM.
Keywords: Juvenile dermatomyositis; Heliotrope rash; Gottron papules; Proximal muscle weakness; Muscle inflammation
| Introduction | ▴Top |
Juvenile dermatomyositis (JDM) is a rare systemic autoimmune vasculopathy affecting approximately 3.2 million children per year in the United States, with 25% of patients younger than 4 years old at disease onset [1]. It is defined by symmetrical proximal muscle weakness, elevated muscle enzymes, and a characteristic violaceous rash over the eyelids with erythematous papules on the extensor joint surfaces [2]. Current diagnostic measures for JDM stem from clinical and laboratory criteria established by Bohan and Peter in 1975, which categorize dermatomyositis as definite, probable, and possible [3]. The five criteria for diagnosis are symmetrical proximal muscle weakness, dermatomyositis associated skin rash (heliotrope rash or Gottron’s sign/papules), elevated serum skeletal muscle enzymes, myopathic electromyography (EMG) pattern, and muscle biopsy with characteristic pathological changes. Definite, probable, and possible dermatomyositis subcategories all revolve around the presence of a characteristic skin rash plus three, two, or one additional criteria, respectively [2, 3]. The noticeable pattern of this diagnostic system is the skin rash requirement, likely secondary to findings that most children with muscle weakness but no rash frequently have in other myopathy types. Although there are other auxiliary findings that can help to rule in myopathy, such as myositis associated or specific antibodies, they are very non-specific, and have only been identified in approximately 40% of pediatric patients [1].
In this case report we examined an atypical presentation and disease course of a 2 years old toddler ultimately diagnosed with JDM, which demonstrated the importance of developing better diagnostic criteria and alternative diagnostic modalities for future patients. It is estimated that characteristic rashes are the first symptom of disease to be recognized in 50% of children with JDM; of those children that do not present with rash, the majority either developed a rash by the time of diagnosis or are diagnosed with an alternate disease process [1, 2]. In the case of our patient, the earliest manifestation of cutaneous findings was 11 months after her initial onset of progressive muscle weakness. At the onset of rash, her muscle weakness had significantly improved solely with rehydration therapy. Up to this point, there had not yet been treatment with any dedicated steroids, methotrexate, or intravenous immunoglobulin (IVIG) therapy. Our patient’s diagnosis therefore came solely on the grounds of confirmatory muscle biopsy in combination with her clinical disease course, and lab work. Although muscle biopsy is a part of the Bohan and Peter diagnostic criteria, a greater number of pediatric rheumatologists are gradually moving away from this procedure due to its invasive nature, with an international survey showing 61% use for diagnostic purposes [1]. Although magnetic resonance imaging (MRI) has recently been added to the revised criteria as an additional standard of measure to help aid in diagnosis, it is only valuable in localizing muscle edema, and cannot establish a cause and therefore confirm a diagnosis [1, 4-6]. Studies have shown that early, aggressive treatment has reduced the risk of disease related morbidity and mortality, but in order to treat we must first be able to diagnose correctly and have adequate tools at our disposal to do so.
| Case Report | ▴Top |
A previously healthy 2 years and 11 months old Caucasian female with unremarkable birth and past medical history, presented with progressive muscle weakness and bilateral knee pain over 7-month duration. She was evaluated in a remote, local facility (Facility A) with limited inpatient resources.
Upon arrival, the patient was described as lethargic, and non-ambulatory in the setting of new onset urinary incontinence, gagging with oral intake, and nasalization of speech. Initial workup revealed an elevated creatinine kinase (CK) and liver transaminases (LFTs) above norm for age [4]. She was subsequently transferred to Facility B for higher echelon of care with greater resources. The patient arrived at Facility B with an admission diagnosis of rhabdomyolysis of unknown etiology. As the patient continued to receive intravenous fluids (IVF), her CK levels began to trend down, but upon discontinuation of fluids, her creatinine spiked once again, prompting her transfer to Facility C. Of note, family was already planning to move to area in which Facility C is located.
On admission to Facility C she exhibited hyperaesthetic bilateral upper and lower extremities, cessation of lower extremity movement, decreased speech, and oromotor control with swallowing difficulties. In addition to her aforementioned myalgias and arthralgias, the patient demonstrated loss of motor strength most pronounced in the hip flexors. Her physical exam was otherwise normal, including a benign skin exam with no visible rashes or lesions. Concerns for metabolic myopathies, idiopathic inflammatory myopathies, dystrophinopathies, autoimmune disorders, as well as mixed connective tissue disease influenced the decisions regarding workup (Table 1). Chest X-ray (Fig. 1) was obtained upon admission, evaluated to be normal. Neither antinuclear antibody nor myositis-associated or specific antibodies were identified in our patient (Table 2). Genetic send out testing was also pursued, and included a chromosomal microarray, myopathy-rhabdomyolysis panel, and muscular dystrophy panel. Further evaluation with lower extremity MRI (Fig. 2) and swallow studies revealed evidence of bilateral myositis and reflux with no aspiration, respectively. A nasogastric tube was placed secondary to poor oral intake, hydration, and for maintenance of CK level, and placement was confirmed by KUB. Over 3 weeks, she slowly regained functions with normalization of speech, and baseline ambulation, although she was still unable to run at discharge.
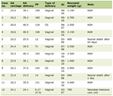 Click to view | Table 1. Laboratory Results for the Patient During Admission to Facility C |
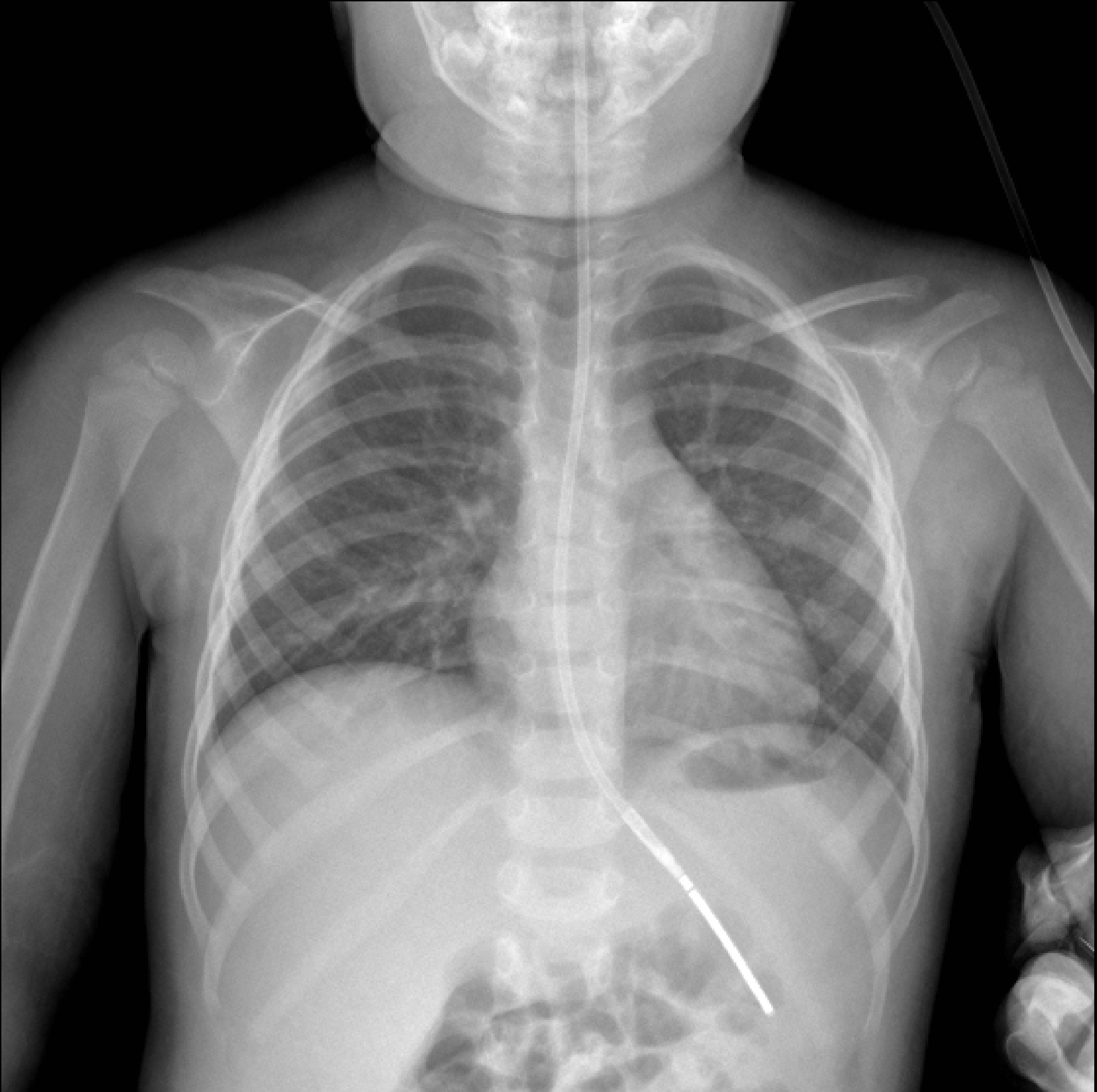 Click for large image | Figure 1. Chest X-ray obtained on admission with unremarkable findings. |
Click to view | Table 2. Laboratory Results for the Patient |
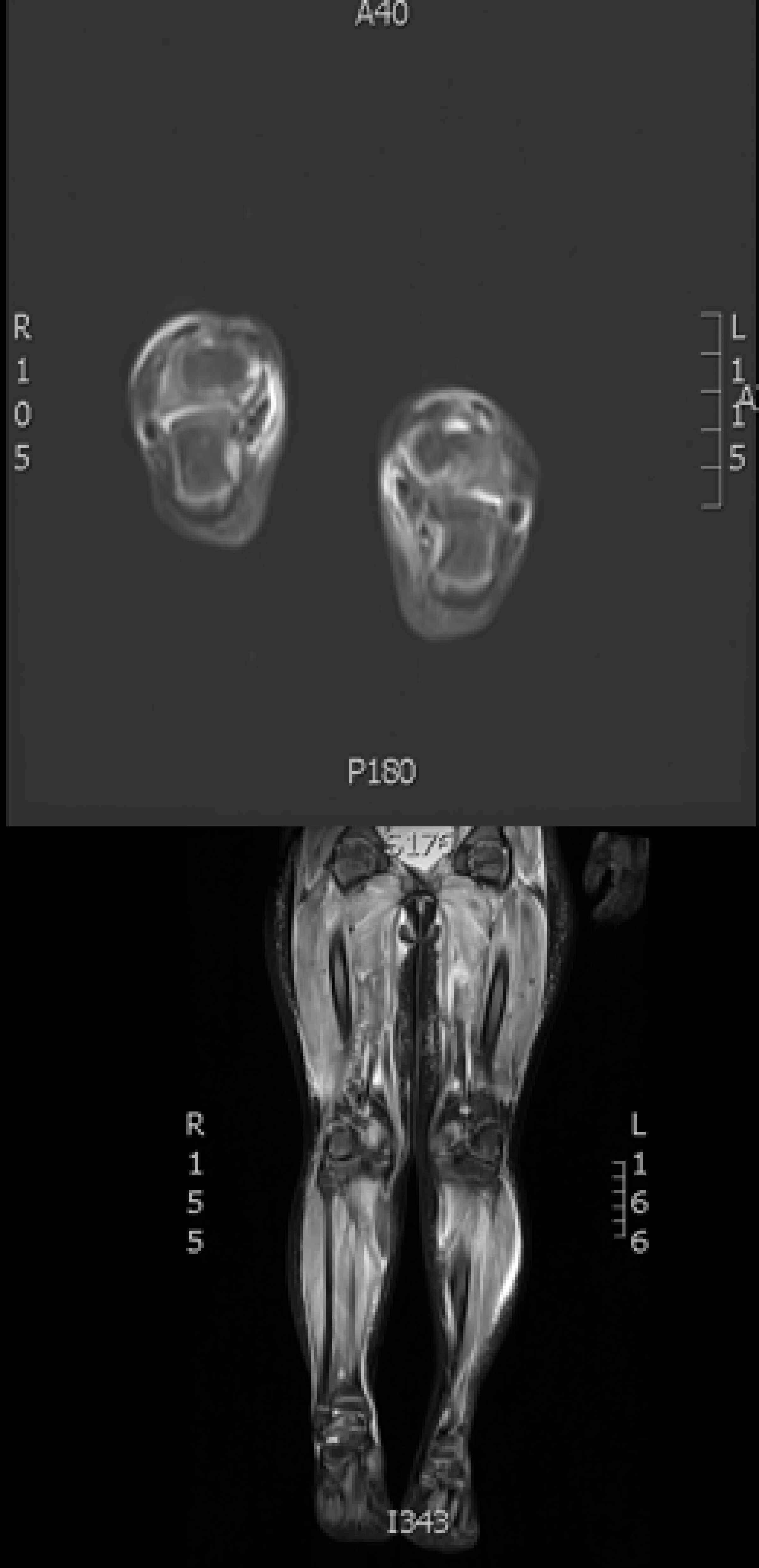 Click for large image | Figure 2. MRI T2-weighted image showing increased signal activity and edema in patient’s proximal and distal lower extremities. |
Approximately 1 month after discharge, genetic and metabolic evaluations were negative for muscular dystrophy, with two variants of unknown significance found on chromosomal microarray (Table 2). Patient was referred to Facility D for further investigation with EMG and muscle biopsy as well as gastrostomy tube placement with Nissen fundoplication. EMG revealed evidence of myopathy, and her muscle biopsy revealed “inflammatory myopathy with fascicular atrophy in both reticular endothelial inclusions,” (Fig. 3). Thus, the patient was suffering from JDM. Up until this point, over 9 months had passed since the onset of muscular and neurologic symptoms without any evidence of cutaneous findings.
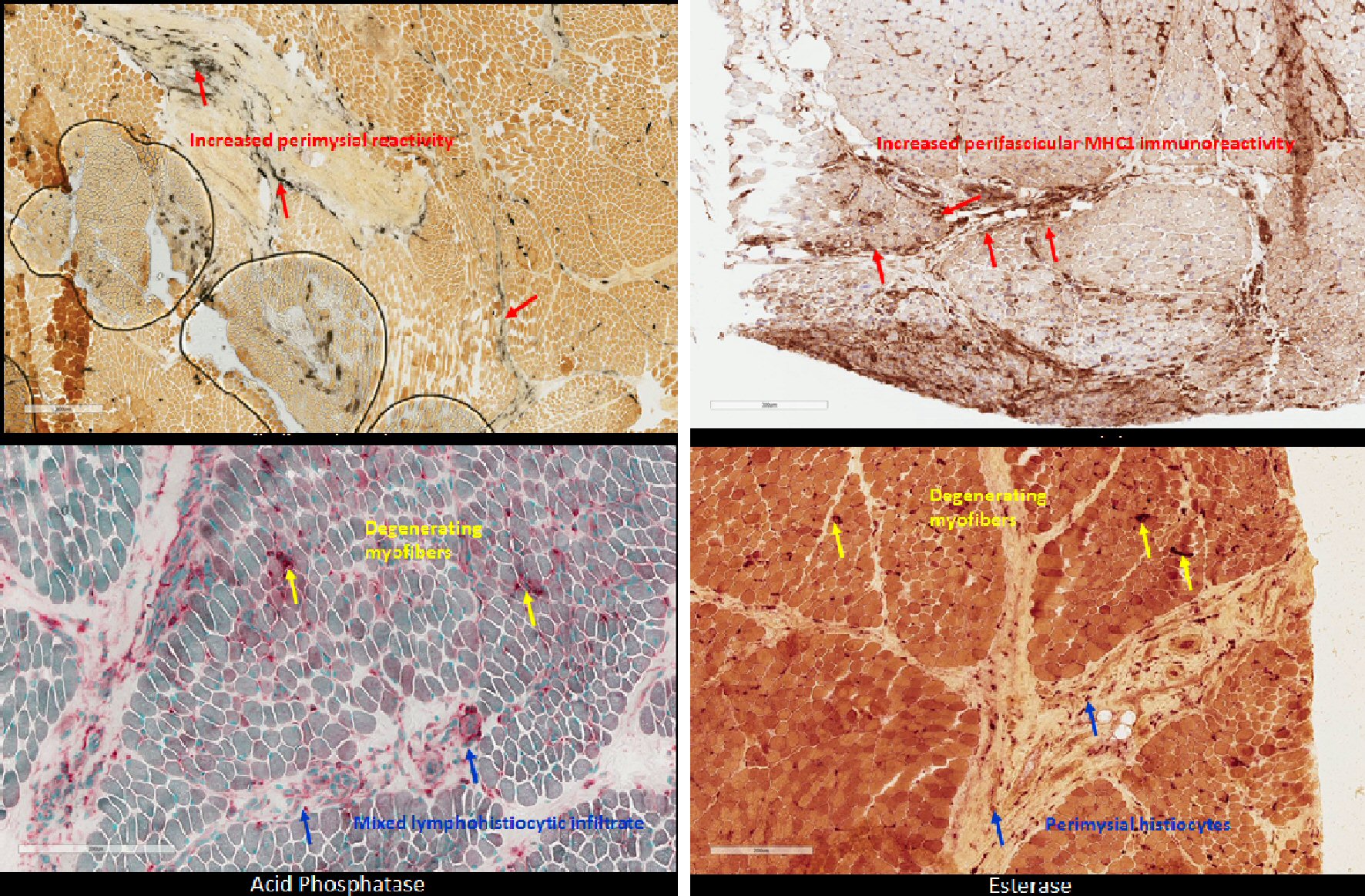 Click for large image | Figure 3. Patient muscle biopsy with histopathology staining demonstrates characteristic perimysial reactivity, perifascicular atrophy, and perifascicular inflammation of lower extremities. |
Over the course of the next 2 months, the patient was evaluated by the rheumatology division at Facility E. During this visit she was found to have a newly developed heliotrope and malar rash, now 11 months since disease onset. Furthermore, the patient had multiple pea sized semi-firm nodules throughout her upper extremity. She subsequently began treatment with weekly methylprednisolone infusions, maintenance prednisolone, methotrexate, folic acid, and vitamin D.
In an attempt to retain the patient within a single hospital system, she was referred to Facility F under the care of a pediatric rheumatologist with a focus in autoimmune myopathies of childhood. Her diagnosis of JDM was again confirmed, she received an initial course of extended duration IVIG, and the case was returned to the child’s Primary Care Manager (PCM) at Facility C.
The care of this patient is ongoing. Through the use of TeleHealth medicine and video teleconference, the PCM from Facility C and the pediatric rheumatologist from Facility F continue to discuss the case and manage the patient’s needs. Currently, she has begun a slow taper of maintenance steroids while maintaining a monthly IVIG infusion regimen and high caloric overnight feeds through G-tube pump for inadequate weight gain.
| Discussion | ▴Top |
JDM is characterized by symmetric proximal muscle weakness, pathognomonic heliotrope and Gottron dermatological manifestations, in addition to elevated CK. These pathognomonic skin rashes have been identified as one of the most important early, diagnostic criteria, as they are appreciated in 88% of children under 18 years old at initial presentation and symptom onset [1, 2, 7, 8]. This disease process results in clinical impairments in multiple activities of daily living that can result in significant morbidity and mortality if diagnosis and intervention are delayed. This patient’s presentation was atypical in that dermatological manifestations were not apparent until 11 months after her initial presentation of bilateral proximal muscle weakness and knee pain, originally causing her to be diagnosed with idiopathic rhabdomyolysis. After a large-scale workup, eventual muscle biopsy showed characteristic histological changes including perimysial reactivity, perifascicular atrophy, perifascicular inflammation, edematous capillary endothelium, and muscle degeneration, clinching the diagnosis. Limitations to our study was the delay of obtaining CBC, aldolase and LDH, secondary to the patient originally having normal labs except for isolated CK elevation at presenting facility. Aldolase and LDH, although not specific, are muscle enzymes known to be elevated in inflammatory myopathies, and could have better guided the diagnostic process if obtained earlier.
JDM diagnostic criteria continue to evolve from the standards developed by Bohan and Peter in 1975 [1-3, 7]. These criteria primarily consist of evidenced increase in activity of specific muscle enzymes in the serum, symmetric proximal muscle weakness, at least one characteristic skin rash, characteristic EMG and muscle biopsy findings. One of the major issues with this criterion is the requirement of at least one of the characteristic skin rashes to have probable JDM. Although majority of children do present with skin rash, a recent study at Nationwide Children’s Hospital revealed that 21% of children under 3 years of age at onset of symptoms, had no rash at diagnosis, as was seen in our patient’s presentation [7]. As our case demonstrates, a delayed cutaneous finding may delay acquisition of muscle biopsy to establish definitive diagnosis.
Another finding that insinuates the need for revised criteria is the use of alternate techniques, such as MRI, to evade the use of more invasive techniques such as muscle biopsy and EMG. When surveyed internationally, use of EMG and muscle biopsy for diagnosis by pediatric rheumatology was 56% and 61%, respectively [1]. Although MRI is a very sensitive approach to gauging and localizing muscle inflammation, it comes at the expense of decreased specificity when compared with muscle biopsy and EMG. MRI can be an excellent tool for less complicated and atypical cases when used in the broader scheme of clinical findings. Many prominent pediatric facilities have used MRI as a part of the “modified Bohan and Peter” criteria to aid in diagnosis when clinical evidence for JDM exists and families or providers which to forgo the muscle biopsy or EMG findings are equivocal [5-7]. Although MRI has been an additional asset in the assessment and diagnosis of JDM, its inability to specifically distinguish between inflammatory disease processes proves to be a limiting factor.
An additional limiting factor in diagnosis for children is the paucity of identifiable autoantibodies, which have only been recognized in up to 40% of children with idiopathic inflammatory myopathies [1]. As was seen in our patient, more than half of children test negative for myositis-associated and specific antibodies such as anti-mi-2, anti-signal recognition particle and anti-p155. These findings therefore showcase the necessity for further research to identify additional serum markers that are more prominent and specific for JDM. The identification of these markers in combination with expanded diagnostic criteria, to include probable JDM for children without rash, would improve early diagnosis and outcomes for children in the future. Further study into the use of blood-borne IL-6, IL-17, IL-23, IFN-β, BAFF, resistin, TLR3 and TLR-7 may emerge as potential biomarkers for the diagnosis of JDM [9].
Conclusions
The case represented a delayed onset of cutaneous findings in a patient with muscle biopsy-proven JDM. Despite access to and expert care coordination, the time course from initial evaluation to obtaining the muscle biopsy was prolonged and highlights the difficulty with establishing diagnosis with this pathology. The care of this patient is complex, and our team would posit that further investigation into the diagnosis and management of JDM is warranted. Finally, our case outlines that clinical suspicion for JDM even in the absence of cutaneous manifestations should warrant urgent evaluation and referral to a skilled pediatric rheumatologist.
Conflict of Interest
Salary support was provided for Drs. Antoine, Reeves, Rohena, Jones, Faux by the United States Department of Defense. The authors have no financial relationships relevant to this article to disclose.
Disclosure
This work was prepared as part of the official duties of Drs. Antoine, Reeves, Rohena, Jones, Faux who are employed by the United States Air Force and Army. The views expressed in this article are those of the authors and do not reflect the official policy or position of the United States Army, Air Force, Department of Defense, or the United States Government.
Funding Support
There was no funding support beyond the salary disclaimers, as above, for the completion of this project.
Author Contributions
Maya Antoine contributed to study design, developed the data analysis plan, interpreted the data, and revised the manuscript. Patrick Reeves provided direct oversight for case description, provided care to the patient, revised the manuscript and provided clinical insight and expertise. Luis Rohena revised the manuscript and provided clinical insight and expertise. Olcay Jones revised the manuscript and provided clinical insight and expertise. Brian Faux drafted the manuscript, and approved the final manuscript, and served as project manager. All authors approved the final version of the manuscript.
| References | ▴Top |
- Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371(9631):2201-2212.
doi - McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, Pilkington CA, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland) - clinical characteristics of children recruited within the first 5 yr. Rheumatology (Oxford). 2006;45(10):1255-1260.
doi pubmed - Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344-347.
doi pubmed - Tschudy MM, Arcara KM, eds. The Harriet Lane Handbook: A Manual For Pediatric House Officers. Philadelphia, PA: Mosby Elsevier, 2012.
- Maillard SM, Jones R, Owens C, Pilkington C, Woo P, Wedderburn LR, Murray KJ. Quantitative assessment of MRI T2 relaxation time of thigh muscles in juvenile dermatomyositis. Rheumatology (Oxford). 2004;43(5):603-608.
doi pubmed - Keim DR, Hernandez RJ, Sullivan DB. Serial magnetic resonance imaging in juvenile dermatomyositis. Arthritis Rheum. 1991;34(12):1580-1584.
doi pubmed - Patwardhan A, Rennebohm R, Dvorchik I, Spencer CH. Is juvenile dermatomyositis a different disease in children up to three years of age at onset than in children above three years at onset? A retrospective review of 23 years of a single center's experience. Pediatr Rheumatol Online J. 2012;10(1):34.
doi pubmed - Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, Miller FW, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013;92(1):25-41.
doi pubmed - Olazagasti JM, Niewold TB, Reed AM. Immunological biomarkers in dermatomyositis. Curr Rheumatol Rep. 2015;17(11):68.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.