

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Case Report
Volume 9, Number 3, March 2017, pages 216-220
Atypical Teratoid/Rhabdoid Tumor of the Sellar Region in an Adult With Long Survival: Case Report and Review of the Literature
Mussa Hussain Almalkia, b, f, Ashjan Alrogia, Abdulkarim Al-Rabiec, Sadeq Al-Dandanb, d, Abdullah Altwairgie, Yasser Orzc
aObesity, Endocrine, and Metabolism Center, King Fahad Medical City, Riyadh, Saudi Arabia
bKing Fahad Medical City, College of Medicine, King Saud bin Abdulaziz University for Health Science, Riyadh, Saudi Arabia
cDepartment of Neurosurgery, National Neuroscience Institute, King Fahad Medical City, Riyadh, Saudi Arabia
dDepartment of Anatomical Pathology, King Fahad Medical City, Riyadh, Saudi Arabia
eOncology Center, King Fahad Medical City, Riyadh, Saudi Arabia
fCorresponding Author: Mussa Hussain Almalki, Obesity, Endocrine, and Metabolism Center, King Fahad Medical City, Riyadh, Saudi Arabia
Manuscript accepted for publication January 17, 2017
Short title: Atypical Teratoid/Rhabdoid Tumor
doi: https://doi.org/10.14740/jocmr2922w
| Abstract | ▴Top |
Atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant central nervous system neoplasm usually diagnosed in young children, although it can occur in adults. Prognosis for AT/RT is poor, with a median survival of 10 - 11 months. We report a rare case of adult sellar and suprasellar AT/RT in a 36-year-old female patient. She was treated with multi-modalities including surgery, chemotherapy and radiation. She markedly improved following treatment with no recurrence in 3 years follow-up. To our knowledge, this is the 11th case of an adult-onset AT/RT in the sellar or suprasellar region with favorable long-term outcome.
Keywords: Atypical teratoid/rhabdoid tumor; Sellar/suprasellar lesion; Radiotherapy; Chemotherapy; Surgery
| Introduction | ▴Top |
Atypical teratoid/rhabdoid tumor (AT/RT) is a very aggressive central nervous system (CNS) neoplasm that is most often seen in infants and young children [1, 2]. About half of AT/RTs are found in the posterior fossa but can also occur anywhere in the brain or spinal cord. It was originally described a histological variant of Wilms’ tumor in 1978 [3]. Primary intracranial diseases were initially reported in 1987 and subsequently, defined as a distinct CNS neoplasm in 1996 and added to the World Health Organization (WHO) Brain Tumor Classification in 2000 (grade IV) [1, 4, 5].
Prognosis for AT/RT is poor, with a median survival time of less than 1 year [1-3]. The tumor is characterized by loss of function mutations in INI-1/hSNF5 gene located on chromosome 22q11.2 [6]. The clinical presentation varies with tumor location. Typically, patient with AT/RT is treated with surgery and craniospinal radiation therapy which is often followed by systemic chemotherapy. Here we report a case of AT/RT that originated in sellar and suprasellar region in a 36-year-old female patient with brief review of the survival rate in this rare aggressive tumor in adult population.
| Case Report | ▴Top |
A 36-year-old female not known to have any medical illnesses, mother of five children presented to the emergency department on October 2013 with a history of severe headache associated with double vision and vomiting.
The headache was progressive over 3 months, then became severe and associated with vomiting and double vision 1 month prior to presentation. She denied galactorrhea, but her menstruation had ceased 2 months earlier. There was no weakness or convulsion. She was on no medication, and had no significant past medical history and family history was unremarkable. Her neurological examination showed no neurological deficits apart from bilateral sixth nerve palsies with pale optic disc. The rest of examination was unremarkable.
Hormone profiles including cortisol, plasma adrenocorticotropic hormone (ACTH), thyroid hormones, follicle-stimulating hormone (FSH), luteinizing hormone (LH) and insulin-like growth factor-1 (IGF-1) were all low as a result of panhypopituitarism (Table 1). In fact, FSH was at 0.5 IU/L, LH was at 0.02 IU/L, FT4 was low at 8 pmol/L with an inadequate level of thyroid-stimulating hormone (TSH) at < 0.005 mIU/L and growth hormone (GH) was low at 0.5 mIU/L. Her basal serum cortisol (measured at 8 a.m.) was 16 nmol/L and cortisol measurement after a synacthen stimulation test was 50 and 85 nmol/L at 30 and 60 min, respectively. She received replacement therapy with L-thyroxine 100 μg/day and hydrocortisone 10 mg a.m. and 5 mg p.m.
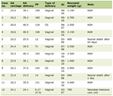 Click to view | Table 1. Serial Pituitary Hormone Profile |
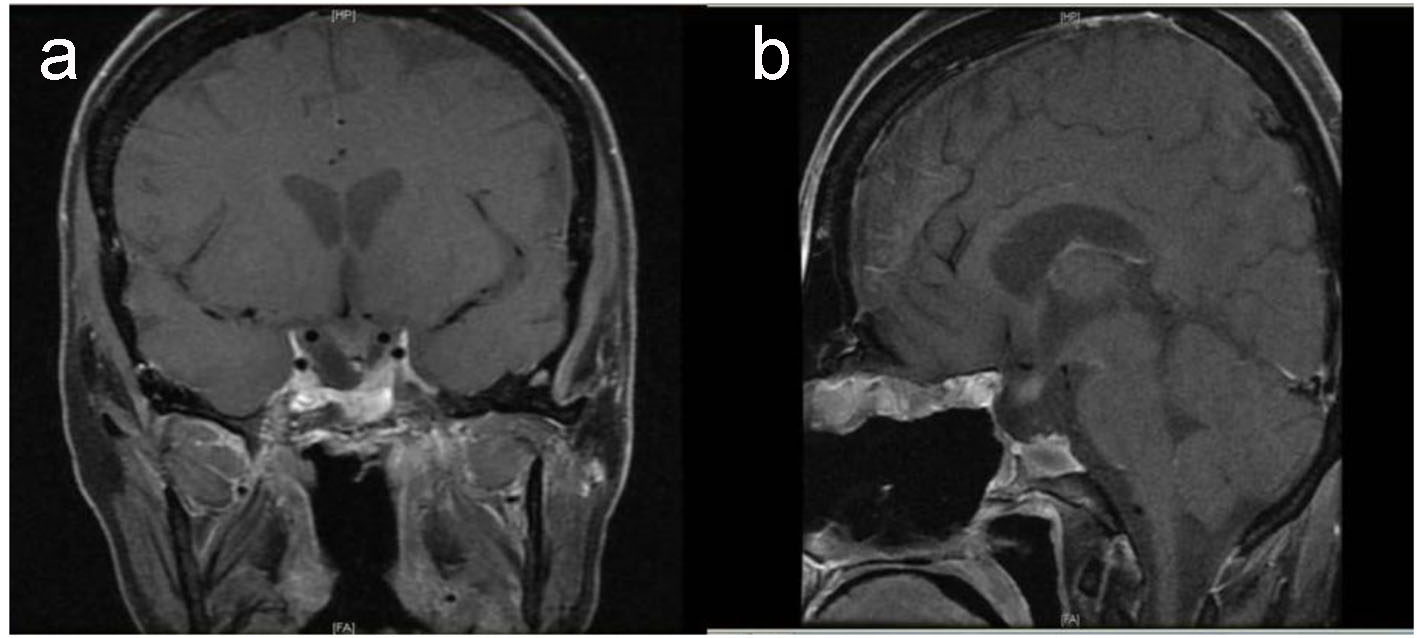
Preoperative magnetic resonance imaging (MRI) identified a large sellar mass with suprasellar extension (Fig. 1).
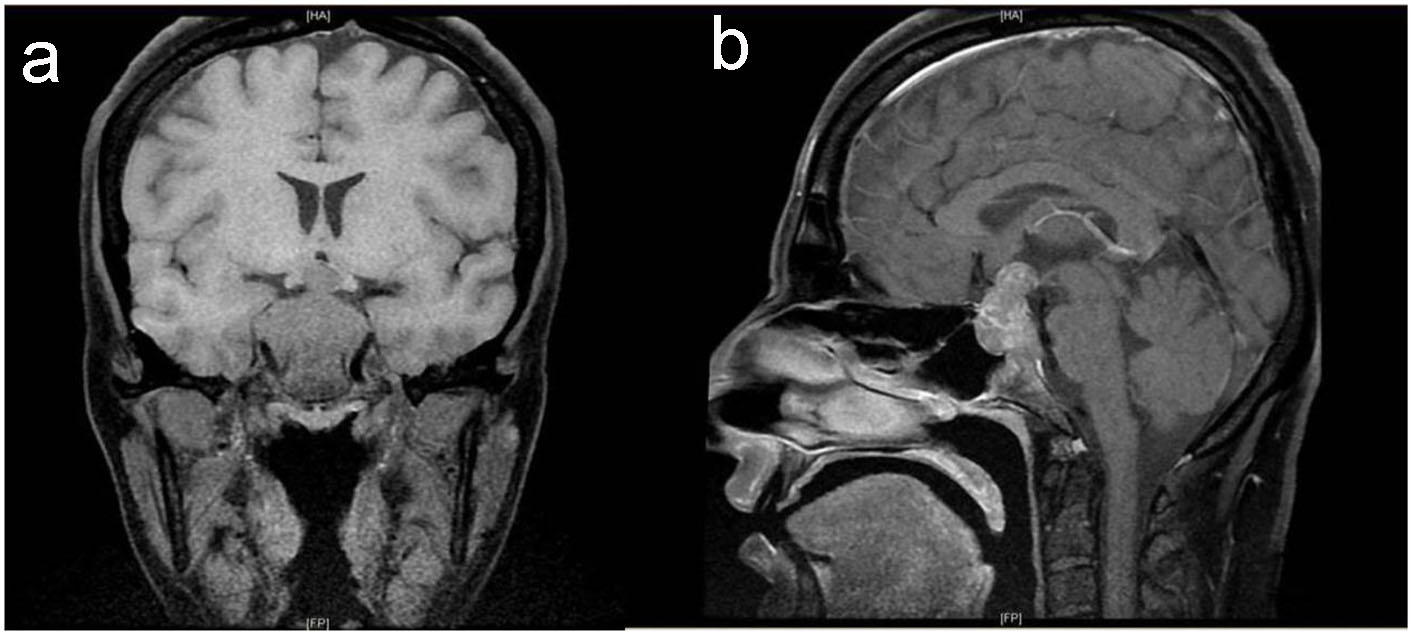 Click for large image | Figure 1. Baseline pituitary MRI. T1-weighted MRI coronal (a) and sagittal (b) images show large intrasellar mass with suprasellar extension compressing and displacing the optic chiasm with evidence of tumor ischemia, bilateral invasion of cavernous sinus and clivus with posterior destruction of clinoid. |
Transphenoidal approach and tumor resection was performed on November 2013 with subtotal resection of the sellar and suprasellar lesion. Postoperatively, the patient’s neurological status remained unchanged. She developed diabetes insipidus that was treated with desmopressin, and continued to require cortisol and thyroxin and discharged without complication.
The histopathological study revealed a high-grade densely cellular neoplasm with frequent foci of necrosis (Fig. 2). Tumor cells were large-sized, polygonal in shape and arranged in sheets. The cytoplasm was clear to eosinophilic with focal eccentric eosinophilic globular inclusions. The nuclei were oval-shaped and pleomorphic with prominent nucleoli (Fig. 3). Immunohistochemical stains for cytokeratins were all negative ruling out metastatic carcinomas (CK (AE1/AE3), CK7, CK20, Cam 5.2 and CK5/6). Mesenchymal markers were all negative apart from vimentin (desmin, SMA, myogenin, and MyoD1). Immunostains for germ cell tumors were all negative (AFP, B-hCG, PLAP, and CD117). Neuronal as well as glial markers were negative as well excluding neuroendocrine tumors, primitive neuroectodermal tumors and gliomas (synaptophysin, chromogranin, and GFAP). Immunostains for melanoma were non-reactive (S100, melan-A, and HMB45). All immunostains for pituitary hormones were negative (GH, prolactin, TSH, FSH, LH, and ACTH). The tumor nuclei were negative for INI-1 (BAF 47) (Fig. 4) and positive for cyclin D1. The morphologic characteristics and immunoprofile were diagnostic of AT/RT.
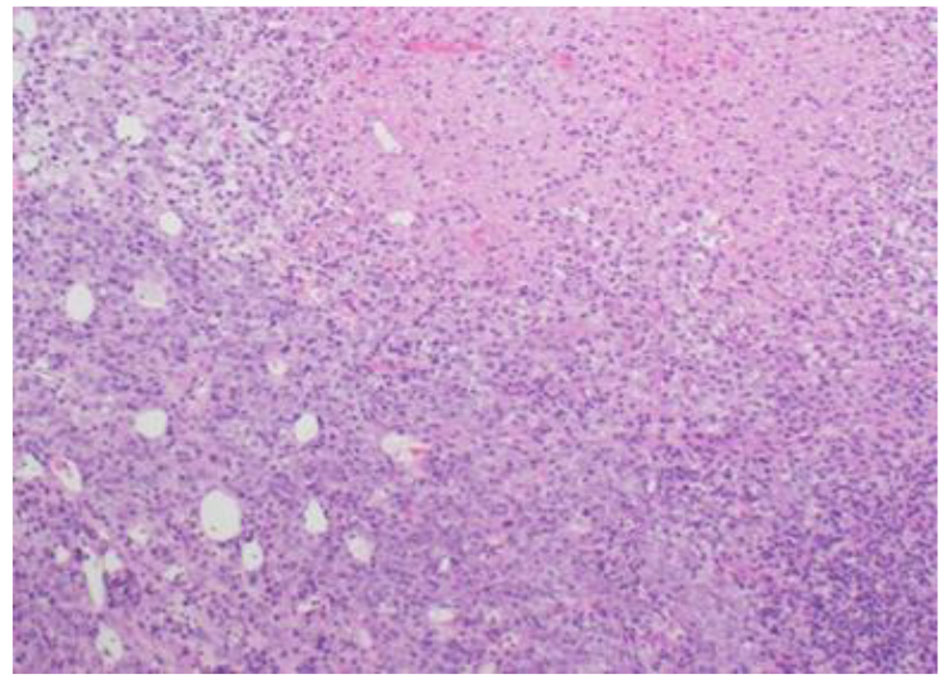 Click for large image | Figure 2. Tumor is composed of sheets of undifferentiated cells with large area of necrosis (H&E stain, ×100 magnifications). |
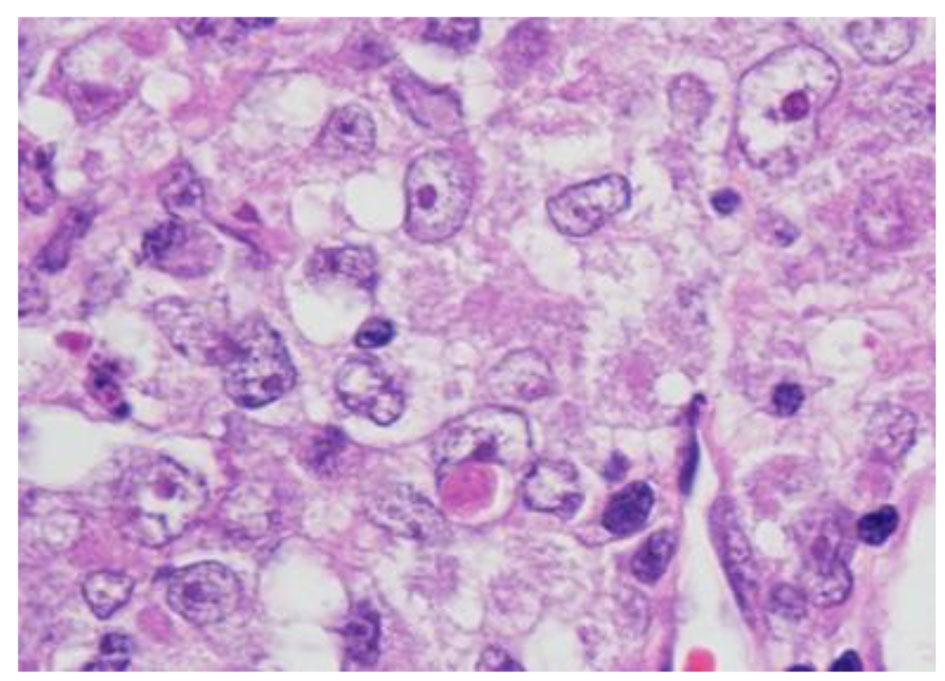 Click for large image | Figure 3. Tumor cells have oval nuclei and prominent nucleoli with focal eosinophilic globular inclusions (H&E stain, × 1,000 magnification with oil). |
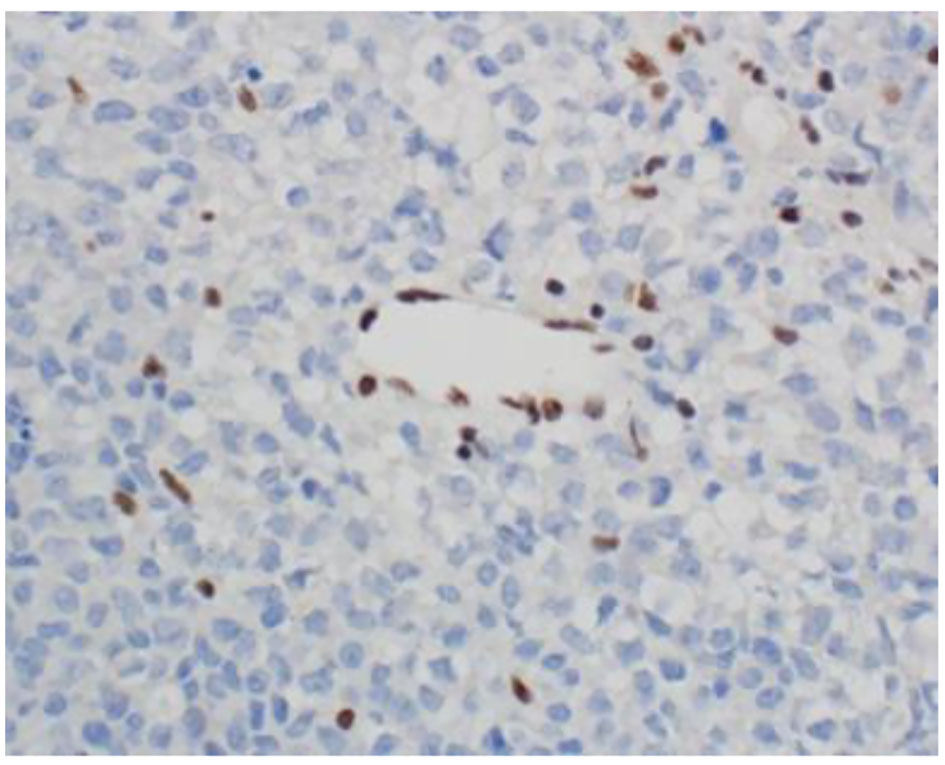 Click for large image | Figure 4. INI-1 (BAF47) immunostain shows loss of nuclear staining in the tumor nuclei and retention of nuclear staining in the lymphocytes and endothelial cells (× 400 magnification). |
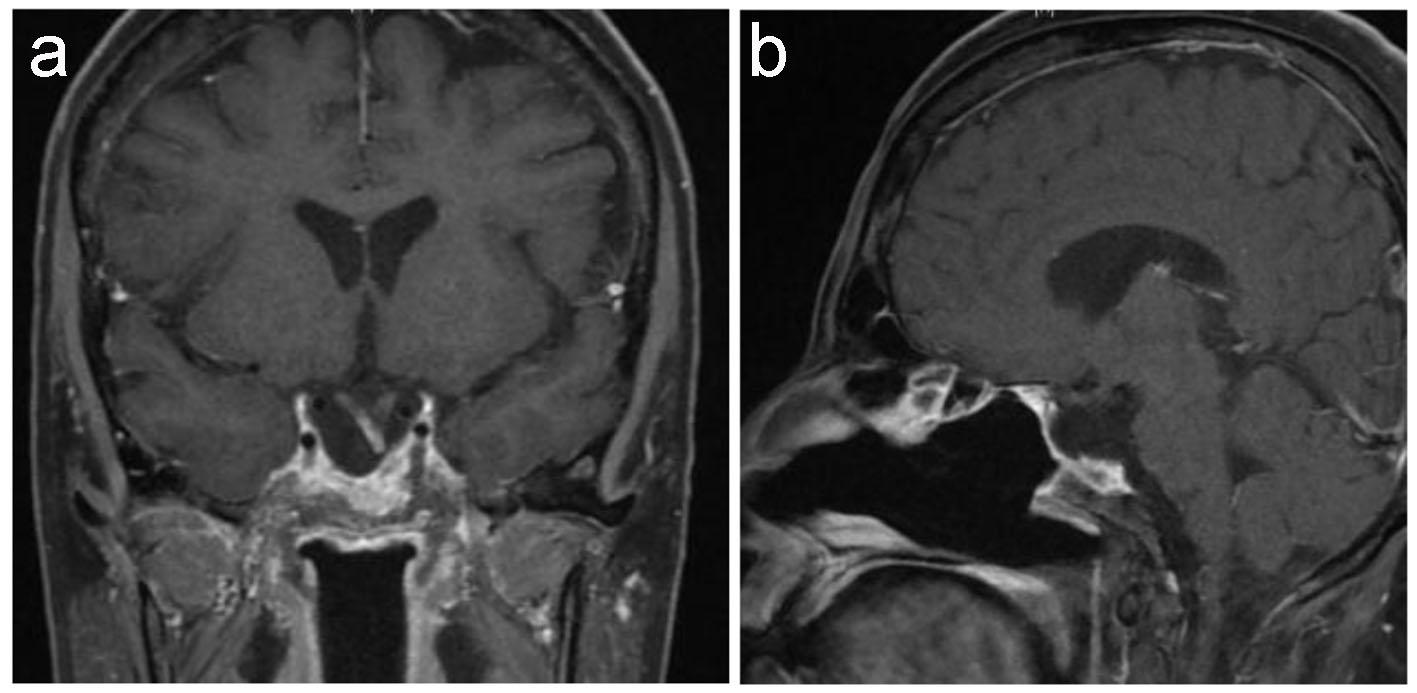
Given the clinically aggressive natural history of this tumor, the patient was referred to oncology center, where she immediately commenced on concurrent chemotherapy (vincristine) with radiotherapy 60 Gy in 30 fractions followed by six cycles of chemotherapy; ICE protocol (ifosfamide, carboplatin, and etoposide) without any significant complication. After her sixth cycle of chemotherapy and radiation therapy, her condition improved and the pituitary MRI revealed a small residual tumor (Fig. 5).
 Click for large image | Figure 5. Postoperative pituitary. T1-weighted MRI coronal (a) and sagittal (b) images show small residual tumor affecting left side of sella with evidence of empty sella, stable displaced pituitary stalk and down displaced optic chiasm. |
She has been on regular follow-up visits at the outpatient clinic for the last 3 years with no clinical or radiological evidence of recurrence after which a good recovery was noted, with a eucortisolemic and euthyroid state on hormonal replacement therapy. However, bilateral sixth nerve and optic atrophy had persisted.
Last pituitary MRI on November 2016 (Fig. 6) demonstrates stable tumor with no evidence of new appearing enhancing lesion to suggest any recurring tumor.
 Click for large image | Figure 6. Latest pituitary MRI. T1-weighted MRI coronal (a) and sagittal (b) images show stable empty sella. Stable deformed left-sided displaced pituitary stalk as well as of down displacement of the optic chiasm and the floor of the third ventricle with focal enhancement of the optic chiasm remains unchanged since the previous study. |
| Discussion | ▴Top |
AT/RT is a rare, fast-growing tumor usually diagnosed in infants and young children who usually die within 1 year after the diagnosis despite aggressive therapy, although it can occur in adults who usually have better prognosis with some longer-term survivors [7]. In 1992, Horn et al [8] were the first authors to recognize AT/RT in an adult patient. Only 45 cases of adults with AT/RT have been reported in the literature to date [9]. AT/RT accounts for approximately 1-2% of pediatric cancers of the CNS [9]. It can occur anywhere in the CNS including the spinal cord. The majority of these tumors (approximately 60%) occur in the posterior cranial fossa (particularly the cerebellum). Its occurrence in the sellar region is rare, with only 11 cases reported in the literature to date including our case (Table 2) [10-17].
Click to view | Table 2. Summary of Previously Described Cases of Sellar AT/RT and the Present Case |
Typically, AT/RTs have a male predominance, with a reported ratio of 3:2 to 2:1 (M/F) [2, 18], although female predominance is notable for sellar region [10].
Patients with sellar AT/RT present with non-specific symptoms, like headache and visual symptoms, which are indistinguishable from that of non-functioning pituitary adenoma. Moreover, the pituitary hormone deficiency is seen in our patient also typically seen in patients with large non-functioning pituitary adenoma with no discriminating endocrine findings to differentiate AT/RTs from pituitary adenoma [19].
Radiological findings of sellar AT/RT are non-specific but are remarkably similar to pituitary adenoma; the lesions are isointense on T1-weighted imaging and enhanced following gadolinium administration [11]. The diagnosis of AT/RT is predominantly based on the tumor’s morphologic and immunohistochemical features. AT/RTs are characterized by rhabdoid cells and loss of INI-1 nuclear immunostaining due to mutations of the INI-1/hSNF5 gene [20, 21]. Decisions on treatment in adults are extrapolated from the pediatric literature. The mainstays in the treatment of AT/RT include surgery, chemotherapy, and radiotherapy [22, 23].
The best chemotherapeutic regimen for treatment of AT/RT is still unknown but the most commonly used regimens are ifosfamide, carboplatin, and etoposide [24]. Patients were treated with chemotherapy, and had survived longer than patients treated with only surgery and irradiation. This could suggest that adult AT/RT cases may have a different biological nature in comparison with most pediatric cases [7].
In our case, aggressive resection followed by multimodality treatment yielded long-term survival with no evidence of recurrence. Of the 10 adult patients reported in the literature (Table 2), four patients survived more than 12 months and our patient survived for 37 months after the diagnosis and complete resection of tumors and adjuvant multimodal treatment.
Conclusion
We reported a case of sellar and suprasellar AT/RT in an adult female who was successfully treated with surgery, radiotherapy and chemotherapy with better prognosis and survival of 37 months. Given the rarity of the disease, AT/RT should be considered in the differential diagnosis of malignant sellar lesion in adult patients.
Funding
This study was not funded.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethical Approval
This article does not contain any studies with human participants performed by any of the authors.
Consent
Written consent was obtained from the patient to reproduce information appearing in this work.
| References | ▴Top |
- Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85(1):56-65.
doi pubmed - Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, Duffner PK, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998;22(9):1083-1092.
doi pubmed - Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer. 1978;41(5):1937-1948.
doi - Lefkowitz IB, Rorke LB, Packer RJ. Atypical teratoid tumor of infancy: definition of an entity. Ann Neurol. 1987;22:448-449.
- Kleihues P. Pathology and genetics of tumours of the nervous system. IARC Press: Lyon. 2000. ISBN 92 83 22409 4.
- Swensen JJ, Keyser J, Coffin CM, Biegel JA, Viskochil DH, Williams MS. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet. 2009;46(1):68-72.
doi pubmed - Takahashi K, Nishihara H, Katoh M, Yoshinaga T, Mahabir R, Kanno H, Kimura T, et al. Case of atypical teratoid/rhabdoid tumor in an adult, with long survival. Brain Tumor Pathol. 2011;28(1):71-76.
doi pubmed - Horn M, Schlote W, Lerch KD, Steudel WI, Harms D, Thomas E. Malignant rhabdoid tumor: primary intracranial manifestation in an adult. Acta Neuropathol. 1992;83(4):445-448.
doi pubmed - Souki C, Abdel-Rhaman M, Qasem A, Al-Hussaini M. Atypical teratoid rhabdoid tumor in adulthood. Clin Neuropathol. 2014;33(3):245-250.
doi pubmed - Irina Lev, Xuemo Fan, Run Yu. Sellar Atypical Teratoid/Rhabdoid Tumor: Any Preoperative Diagnostic Clues? AACE Clinical Case Reports: Winter. 2015;1(1):e2-e7.
- Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A. Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol. 2005;15(1):23-28.
doi pubmed - Arita K, Sugiyama K, Sano T, Oka H. Atypical teratoid/rhabdoid tumour in sella turcica in an adult. Acta Neurochir (Wien). 2008;150(5):491-495; discussion 496.
doi pubmed - Schneiderhan TM, Beseoglu K, Bergmann M, Neubauer U, Macht S, Hanggi D, Reifenberger G, et al. Sellar atypical teratoid/rhabdoid tumours in adults. Neuropathol Appl Neurobiol. 2011;37(3):326-329.
doi pubmed - Shitara S, Akiyama Y. Atypical teratoid/rhabdoid tumor in sellar turcica in an adult: A case report and review of the literature. Surg Neurol Int. 2014;5:75.
doi pubmed - Moretti C, Lupoi D, Spasaro F, Chioma L, Di Giacinto P, Colicchia M, Frajoli M, et al. Sella turcica atypical teratoid/rhabdoid tumor complicated with lung metastasis in an adult female. Clin Med Insights Case Rep. 2013;6:177-182.
pubmed - Las Heras F, Pritzker KP. Adult variant of atypical teratoid/rhabdoid tumor: immunohistochemical and ultrastructural confirmation of a rare tumor in the sella tursica. Pathol Res Pract. 2010;206(11):788-791.
doi pubmed - Chou SQH, Lo SSM, Wong HN, et al. Atypical teratoid/ rhabdoid tumour in the sella turcica of a female adult. Hong Kong J Radiol. 2013;16:65-68.
doi - Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, Walter AW, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004;22(14):2877-2884.
doi pubmed - Buscariollo DL, Park HS, Roberts KB, Yu JB. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer. 2012;118(17):4212-4219.
doi pubmed - Uno K, Takita J, Yokomori K, Tanaka Y, Ohta S, Shimada H, Gilles FH, et al. Aberrations of the hSNF5/INI1 gene are restricted to malignant rhabdoid tumors or atypical teratoid/rhabdoid tumors in pediatric solid tumors. Genes Chromosomes Cancer. 2002;34(1):33-41.
doi pubmed - Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203-206.
doi pubmed - Gardner SL, Asgharzadeh S, Green A, Horn B, McCowage G, Finlay J. Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer. 2008;51(2):235-240.
doi pubmed - Park HG, Yoon JH, Kim SH, Cho KH, Park HJ, Kim EH. Adult-onset sellar and suprasellar atypical teratoid rhabdoid tumor treated with a multimodal approach: a case report. Brain Tumor Res Treat. 2014;2(2):108-113.
doi pubmed - Shonka NA, Armstrong TS, Prabhu SS, Childress A, Choi S, Langford LA, Gilbert MR. Atypical teratoid/rhabdoid tumors in adults: a case report and treatment-focused review. J Clin Med Res. 2011;3(2):85-92.
doi
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.