

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Case Report
Volume 8, Number 10, October 2016, pages 764-767
An Overlapping Case of Alport Syndrome and Thin Basement Membrane Disease
Mashriq Alganabia, Ahmad Eterb, c
aAvalon University School of Medicine, 122-124 Santa Rosaweg, Willemstad, Curacao
bDivision of Nephrology, Department of Medicine, Raleigh General Hospital, 1710 Harper Rd, Beckley, WV 25801, USA
cCorresponding Author: Ahmad Eter, Division of Nephrology, Department of Medicine, Raleigh General Hospital, 1710 Harper Rd, Beckley, WV 25801, USA
Manuscript accepted for publication August 18, 2016
Short title: Alport Syndrome
doi: http://dx.doi.org/10.14740/jocmr2740w
| Abstract | ▴Top |
We report a case of a 48-year-old male who presented with hematuria of at least 10 years, and has a daughter with hematuria as well. The patient has a history of degenerative hearing loss, decreased vision and cataract formation, but no diabetes, hypertension or proteinuria. A full serology and urology workup was negative for any abnormality. A kidney biopsy for the patient revealed a diagnosis of Alport syndrome but was unable to rule out thin basement membrane disease. The biopsy was inconclusive in making the diagnosis but the patient’s clinical presentation led to the diagnosis of Alport syndrome. The patient’s 10-year-old daughter also has hematuria with no clear etiology but now can subsequently be anticipatorily managed for Alport syndrome progression. Due to the rarity of the disease, diagnosis is often missed or delayed by primary care providers especially when no associated proteinuria has yet developed. This can lead to confusion and misdiagnosis with thin basement membrane disease, a generally benign hematuria without kidney failure progression. Additionally, biopsy can be inconclusive in these patients, relying on the physician’s history and physical examination findings to diagnose. It is important to appropriately diagnose Alport syndrome not only to manage the patient’s rate of kidney failure progression but also allow for a higher degree of suspicion, screening and intervention in the patient’s family members. Both the inconclusive nature of kidney biopsies and the usefulness of diagnosis for family member screening are often overlooked in medical literature but are explored in this case.
Keywords: Hereditary nephritis; Alport syndrome; Thin basement membrane disease; Hematuria; Collagen IV; Progressive glomerulonephritis
| Introduction | ▴Top |
Hereditary nephritis or Alport syndrome is a rare inherited progressive glomerular disease caused by mutations in the genes encoding the alpha-3, alpha-4, and alpha-5 chains of type IV collagen found in various basement membranes including the kidney, eye, and cochlea [1, 2]. Alport syndrome usually presents with hematuria, proteinuria, progressive renal failure, ocular abnormalities, and sensorineural hearing deficits. It is a rare disease, with a prevalence of 1 in 50,000 live births [3]. Alport disease often presents before 10 years of age with gross hematuria and occasionally subsequent to an upper respiratory infection [4]. Males are affected more than females as the primary mode of inheritance is X-linked, but autosomal recessive and autosomal dominant variants exist as well. With time, proteinuria and hypertension develop which lead to progressive renal insufficiency and end-stage kidney disease often between the ages of 16 and 35 years in the X-linked or autosomal recessive variants, but can be more indolent in the autosomal dominant type, leading to renal failure between the ages of 45 and 60 years [1, 5].
Thin basement membrane disease also presents with hematuria and often early in childhood. However, it has a generally autosomal dominant inheritance pattern and tends to be non-progressive with minimal proteinuria and normal renal function [6].
Differentiating between thin basement membrane disease and Alport syndrome can often be made with a kidney biopsy; however, in our case the biopsy was inconclusive.
| Case Report | ▴Top |
We report a case of a 48-year-old Caucasian male who presented with right-sided flank pain and hematuria. The patient described the hematuria as being persistent for greater than 10 years, while the right-sided flank pain was more acute, progressing over the last week, sharp in character, and without radiation. On taking the patient history, there was no notable dysuria, but he did have increased urinary frequency and nocturia. The patient’s history additionally revealed progressive hearing loss over several years along with decreased vision and cataract formation. Notably, the patient did not have type 2 diabetes, hypertension, or proteinuria. Blood work for the patient including complete blood count and a basic metabolic panel was normal, and extensive serology and urology workup to rule out other causes of hematuria was negative. A renal ultrasound showed normal appearing kidneys bilaterally. Based on the clinical presentation of the patient, a kidney biopsy was recommended to which the patient agreed as his 10-year-old daughter had worsening of her hematuria, and if he did have Alport syndrome, his daughter may be able to better manage her disease progression.
The CT scan guided native kidney biopsy for the patient revealed normal glomeruli by light microscopy but showed significant segmental glomerular basement membrane thinning and thickening on electron microscopy (Fig. 1), suggestive of Alport syndrome. Immunofluorescence histology of nine glomeruli showed no segmental or global sclerosis. Collagen IV staining showed preserved linear alpha-5 staining of the glomerular basement membranes and neighboring tissue (Fig. 2), suggesting either normal kidney architecture or thin basement membrane disease.
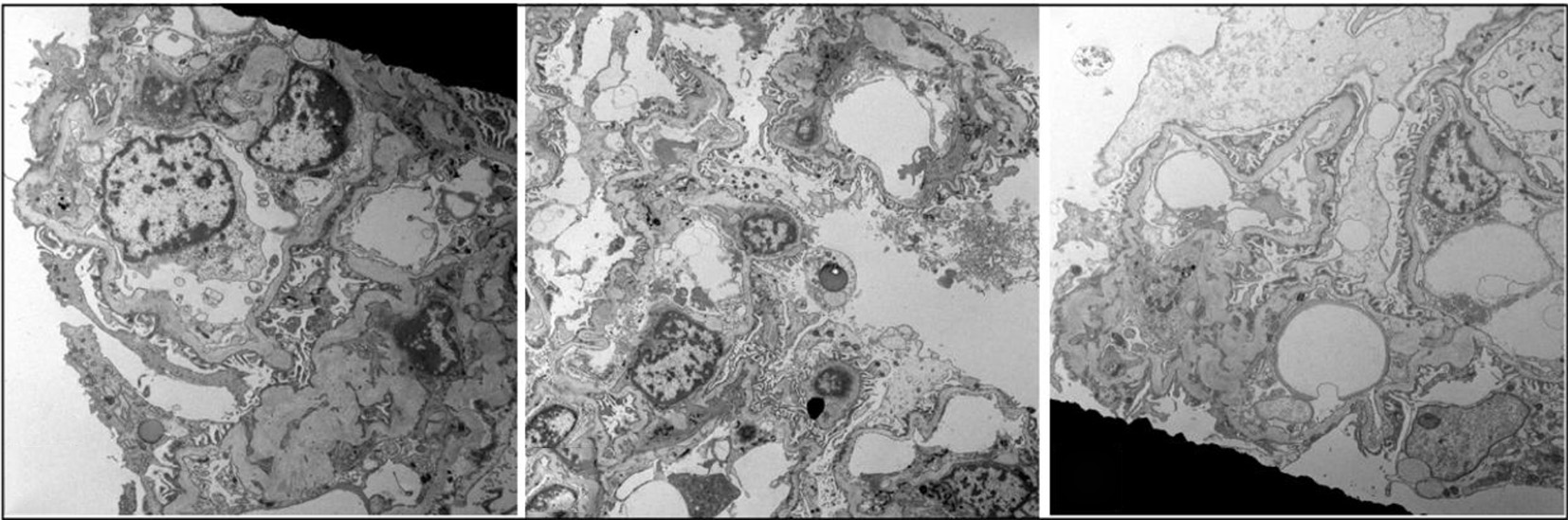 Click for large image | Figure 1. Electron microscopy showing significant segmental glomerular basement membrane thinning alternating with segmental thickening and lamellations suggestive of Alport syndrome. |
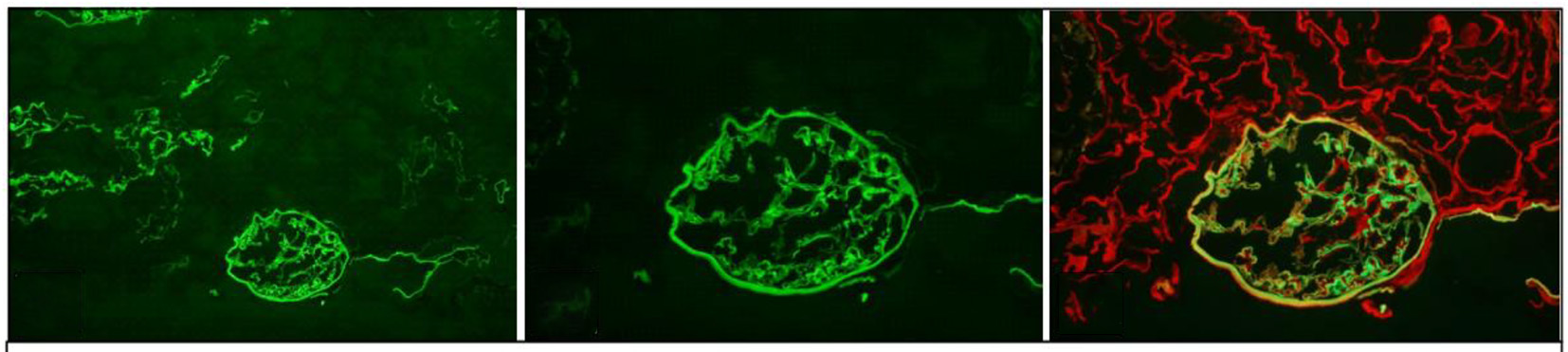 Click for large image | Figure 2. Preserved linear alpha-5 staining of the glomerular basement membranes, Bowman’s capsule and distal tubular basement membranes suggestive of normal architecture or thin basement membrane disease. |
| Discussion | ▴Top |
Hereditary nephritis (or Alport syndrome) is an inherited form of progressive glomerulonephritis. The disease is often associated with ocular and hearing deficits generally before the age of 20 years. In the case of the ocular abnormalities, they include dot-and-fleck retinopathy (most common), anterior lenticonus or posterior polymorphous corneal dystrophy [7]. In addition, patients, especially male, often develop hearing deficits that start in the high frequency range and progress over time to include lower frequencies such as those of conversational speech. Renal manifestations of Alport syndrome often initially start as asymptomatic persistent microscopic hematuria that progresses to episodic gross hematuria. Serum creatinine and blood pressure tend to remain normal in early childhood for these patients, and slowly develop into proteinuria, hypertension and progressive renal insufficiency terminating in end-stage kidney disease generally between the ages of 16 and 35 years [5]. Though female patients generally have less severe manifestations of Alport syndrome, in those with recurrent gross hematuria, proteinuria, and hearing loss, it is associated with more severe renal dysfunction and an earlier end-stage renal disease progression. Additional clinical associations reported with Alport syndrome include leiomyomas and arterial aneurysms of the thoracic and abdominal aorta [8, 9].
Alport syndrome is a rare disease, with an estimated prevalence of about 1 in 50,000 live births and accounts for approximate 3% of children and 0.2% of adults with end-stage renal disease [10]. It arises from mutations in genes encoding the alpha-3, alpha-4, and alpha-5 chains of type IV collagen found in various basement membranes including the kidney, eye and cochlea. Transmission of Alport syndrome can be X-linked, autosomal dominant, or autosomal recessive. X-linked transmission from mutations in the COL4A5 gene on the X chromosome accounts for the majority of patients. Meanwhile, the autosomal dominant variant caused by heterozygous mutations in the COL4A3 and COL4A4 genes accounts for about 25% of Alport patients, and the autosomal recessive variant caused by genetic defects in COL4A3 or COL4A4 genes accounts for the remaining 15% of Alport patients [11].
By contrast, thin basement membrane disease is also an inherited disorder that presents with hematuria, but is often benign without progressive renal insufficiency. Additionally, thin basement membrane disease does not produce proteinuria or have the hearing or ocular deficits often seen with Alport syndrome. It is also far more common with an estimated prevalence of about 1 in 11 - 19 and primarily has an autosomal dominant inheritance pattern [12]. Refer to Table 1 for a brief comparison between thin basement membrane disease and Alport syndrome. Table 2 [13] shows the staining pattern for collagen IV in thin basement membrane disease in comparison to the different variants of Alport syndrome. In Alport syndrome discontinuation of staining in various basement membrane areas including glomerular basement membrane, bowman capsule, and distal tubular basement membrane can often be seen, particularly in the case of the glomerular basement membrane. In the case presented here, the staining showed a preserved linear alpha-5 staining (Fig. 2) which is seen in thin basement membrane but generally not seen in Alport syndrome. However, infrequent exceptions have been noted in the autosomal recessive variant of Alport syndrome [1, 2].
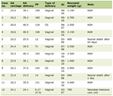 Click to view | Table 1. Alport Syndrome and Thin Basement Membrane Comparison |
Click to view | Table 2. Staining for Alpha-3 and Alpha-5 Chains of Collagen IV in Thin Basement Membrane Disease (TBMD) and Alport Syndrome Variants |
The classical presentation for Alport syndrome is in a young adult or teenage male with hematuria, and some degree of ocular abnormalities and hearing deficits. Additionally, patients tend to have a family history of hematuria associated with proteinuria, renal failure, deafness, and vision problems. This makes evaluation of the patient’s family members for hematuria vital as it may lead to early diagnosis. Early detection of Alport syndrome is very important; although there is no currently available treatment that fully reverses the disorder, there exist multiple methods that help slow down glomerular sclerosis and development of renal failure.
Though immunohistochemistry and immunofluorescence studies to identify the COL4A3, COL4A4, and COL4A5 proteins in the glomerular basement membrane can be helpful in confirming the diagnosis, they can also be normal in some patients with Alport syndrome, especially in those who present with milder variants as was seen in this patient’s case. Consequently, it can lead to a biopsy diagnosis of thin basement membrane disease or normal instead of Alport syndrome. This emphasizes the importance of the clinical presentation as often diagnosis is based on the disease progression and presenting symptoms and not on the biopsy results exclusively. Biopsy results can still often confirm a diagnosis of Alport syndrome but may also be inconclusive as in this case. Symptomatic presentations including hearing deficits, hematuria, and visual abnormalities as well as pertinent family history including family members with Alport syndrome or hematuria and proteinuria are key in helping guide diagnosis and treatment for the patient.
Conclusion
Alport syndrome is a rare inherited form of progressive glomerulonephritis that is often associated with hematuria, ocular and hearing deficits. Unlike thin basement membrane disease, it leads to progressive renal insufficiency and eventual end-stage kidney disease necessitating dialysis. Kidney biopsy can occasionally be non-conclusive and be unable to rule out either a normal kidney or a kidney with thin basement membrane disease as was seen in this case. This further emphasizes the importance of the clinical presentation for diagnosis. Additionally, since both thin basement membrane disease and Alport syndrome have a heavy genetic component, it is important to diagnose patients as early as possible for potential future genetic treatment options as well as increase the index of suspicion and subsequent screening of family members to allow for earlier intervention.
Financial Support
None.
Conflicts of Interest
The authors declare that they have no relevant conflicts of interest.
| References | ▴Top |
- Kashtan CE, Michael AF. Alport syndrome. Kidney Int. 1996;50(5):1445-1463.
doi - Kashtan CE. Familial hematuria due to type IV collagen mutations: Alport syndrome and thin basement membrane nephropathy. Curr Opin Pediatr. 2004;16(2):177-181.
doi pubmed - Levy M, Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 2000;58(3):925-943.
doi pubmed - Gubler M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, Habib R. Alport's syndrome. A report of 58 cases and a review of the literature. Am J Med. 1981;70(3):493-505.
doi - Gubler MC. Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol. 2008;4(1):24-37.
doi pubmed - Aarons I, Smith PS, Davies RA, Woodroffe AJ, Clarkson AR. Thin membrane nephropathy: a clinico-pathological study. Clin Nephrol. 1989;32(4):151-158.
pubmed - Colville DJ, Savige J. Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet. 1997;18(4):161-173.
doi - Dahan K, Heidet L, Zhou J, Mettler G, Leppig KA, Proesmans W, David A, et al. Smooth muscle tumors associated with X-linked Alport syndrome: carrier detection in females. Kidney Int. 1995;48(6):1900-1906.
doi pubmed - Lyons OT, St John ER, Morales JP, Chan YC, Taylor PR. Ruptured thoracoabdominal aortic aneurysm in a renal transplant patient with Alport's syndrome. Ann Vasc Surg. 2007;21(6):816-818.
doi pubmed - Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore). 1999;78(5):338-360.
doi - Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000;11(4):649-657.
pubmed - Dische FE, Anderson VE, Keane SJ, Taube D, Bewick M, Parsons V. Incidence of thin membrane nephropathy: morphometric investigation of a population sample. J Clin Pathol. 1990;43(6):457-460.
doi pubmed - Haas M. Alport syndrome and thin glomerular basement membrane nephropathy: a practical approach to diagnosis. Arch Pathol Lab Med. 2009;133(2):224-232.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.