

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Case Report
Volume 8, Number 9, September 2016, pages 683-687
Acute Onset Anti-Synthetase Syndrome With Pericardial Effusion and Non-Specific Interstitial Pneumonia
Aditya Shaha, b, Samir R. Patela
aAdvocate Christ Medical Center, 4440 W 95th Street, Oak Lawn, IL 60453, USA
bCorresponding Author: Aditya Shah, Advocate Christ Medical Center, 4440 W 95th Street, Oak Lawn, IL 60453, USA
Manuscript accepted for publication July 01, 2016
Short title: Acute Onset Anti-Synthetase Syndrome
doi: http://dx.doi.org/10.14740/jocmr2631w
| Abstract | ▴Top |
Anti-synthetase syndrome (AS) is a clinical entity which is described classically by the triad of interstitial lung disease (ILD), inflammatory myositis and presence of aminoacyl-tRNA synthetase antibodies (ASA). We describe a rare presentation of this condition with regard to the uncharacteristically acute nature of presentation, acute decompensation in clinical condition, development of acute interstitial pneumonitis requiring rescue extracorporeal membrane oxygenation (ECMO) and accompaniment of significant pericardial effusion on presentation, followed by rapid improvement with initiation of steroids.
Keywords: Anti-synthetase syndrome; ECMO; Pericardial effusion
| Introduction | ▴Top |
Anti-synthetase syndrome (AS) is a clinical entity which is described classically by the triad of interstitial lung disease (ILD), inflammatory myositis and presence of aminoacyl-tRNA synthetase antibodies (ASA) [1, 2]. It is often seen with some classic findings like “mechanics hands”, Raynaud’s phenomenon and polyarthritis [3]. The anti-histidyl-tRNA synthetase (anti-Jo-1) antibody is the most commonly reported antibody associated with this condition [1, 2]. Owing to the higher prevalence of lung involvement, the overall prognosis for this syndrome seems to be worse than other myositis disorders. Some studies describe a mortality of close to 70% in patients who have this condition [1]. It is also hypothesized that AS can also result in pulmonary hypertension in about 50% of cases. The clinical spectrum is vast but is usually characterized by chronic progression of shortness of breath, persistent cough, along with crepitations on lung exam with a restrictive pattern of lung disease. The disease, rare as it is, is more prevalent in women than in men. Early diagnosis is challenging, with milder cases being hard to detect. ILD may be the only manifestation of the disease. Severe disease may take time to develop with a possibility of relapses [4]. Radiological findings can range from non-specific interstitial pneumonia (NSIP), characterized by a wide range of radiological presentations: ground glass opacities in peripheral to lower zones; bronchial dilatation and linear opacities; honey combing; or bronchiectasis. It can also present as organizing pneumonia with patchy consolidations and ground glass opacities with multiple nodules or masses. Another possible presentation is as usual interstitial pneumonia characterized by lung inflammation, repair and fibrosis. In some cases, there are also typical findings of reticulation, bronchial dilation and honeycombing with minimal ground glass opacities [5].
| Case Report | ▴Top |
Our patient is a 43-year-old male with no significant past medical history who presented to our emergency room with a chief complaint of shortness of breath for the duration of 2 weeks. Two weeks before presentation, the patient was prescribed azithromycin by his primary care provider for a presumed diagnosis of pneumonia. On further history, the patient endorsed a non-productive cough associated with subjective fevers, chills, weakness and malaise for 1 month. He denied any past medical history of autoimmune, systemic or immune-compromising conditions. He denied any sick contacts, recent travel, tuberculosis or chemical exposure. He denied any nausea, vomiting, diarrhea, muscular weakness or neurological symptoms, orthopnea or paroxysmal nocturnal dyspnea. At baseline, he was able to walk approximately five blocks without getting dyspneic. He had no known drug allergies, took no other medications and his social history was negative other than 20 pack year smoking history. His family history was unremarkable as well. Our patient worked a desk job and his occupational history was negative for any kind of environmental pollutants known to be associated with lung disease. On admission to the emergency room, the patient was afebrile, with a respiratory rate of 26 breaths/min, heart rate of 115 beats/min, with a room air saturation of 75% and blood pressure of 90/60 mm Hg. Physical exam was pertinent for hyperkeratosis over the index fingers and his thumbs bilaterally along with thickening noticed over the metacarpophalangeal joints and proximal interphalangeal joints. The rest of the physical exam was unremarkable. Early resuscitation was initiated and the patient was treated with 6 L of oxygen via nasal cannula and given 30 cc/kg of normal saline for fluid resuscitation with normalization of his blood pressure and heart rate prior to admission to the general medical telemetry unit.
On admission, his labs were remarkable for a white blood cell count of 25,000 units/L, hemoglobin level was 13.5 g/dL and platelet count of 385 × 109/L. On his metabolic panel, the patient’s sodium level was 133 mg/dL with a BUN of 13 mg/dL and creatinine of 0.69 mg/dL, with troponin being 0.09 μg/L and B-type natriuretic peptide being 79 pg/mL. His liver function was remarkable for a normal alkaline phosphatase and bilirubin level with aspartate transaminase and alanine transaminase of 259 units/L and 266 units/L, respectively with a CPK of 7,500 International Units/L. Albumin was measured at 2.4 g/dL with a globulin level of 4.2 g/dL. His arterial blood gas values were remarkable at 7.37/43/52 on 6 L nasal cannula oxygen. His lactic acid was elevated at 2.4 mg/dL. ESR on admission was 35 mm/h, CRP was 24 mg/L and procalcitonin level was 34 ng/mL. ANA showed a speckled patter with 1:320 titers, anti-Jo-1 antibody was also positive at 8 and anti-CCP antibody was positive at 153.
A CT scan of his chest on admission (Fig. 1) showed a pericardial effusion measuring 1.3 cm in thickness. Non-confluent areas of ill defined opacities were also seen in both lungs with denser consolidations and air Broncho-grams mainly in the right middle lobe and both the lower lobes which were thought to be a combination of infiltrates and atelectasis. A chest X-ray (Fig. 2) showed a small right-sided pleural effusion with a patchy right sided opacity. An echocardiogram (ECHO) evaluation revealed a moderate to large pericardial effusion (Fig. 3). A chest X-ray at the end of the course of treatment showed complete resolution of the prior findings (Fig. 4).
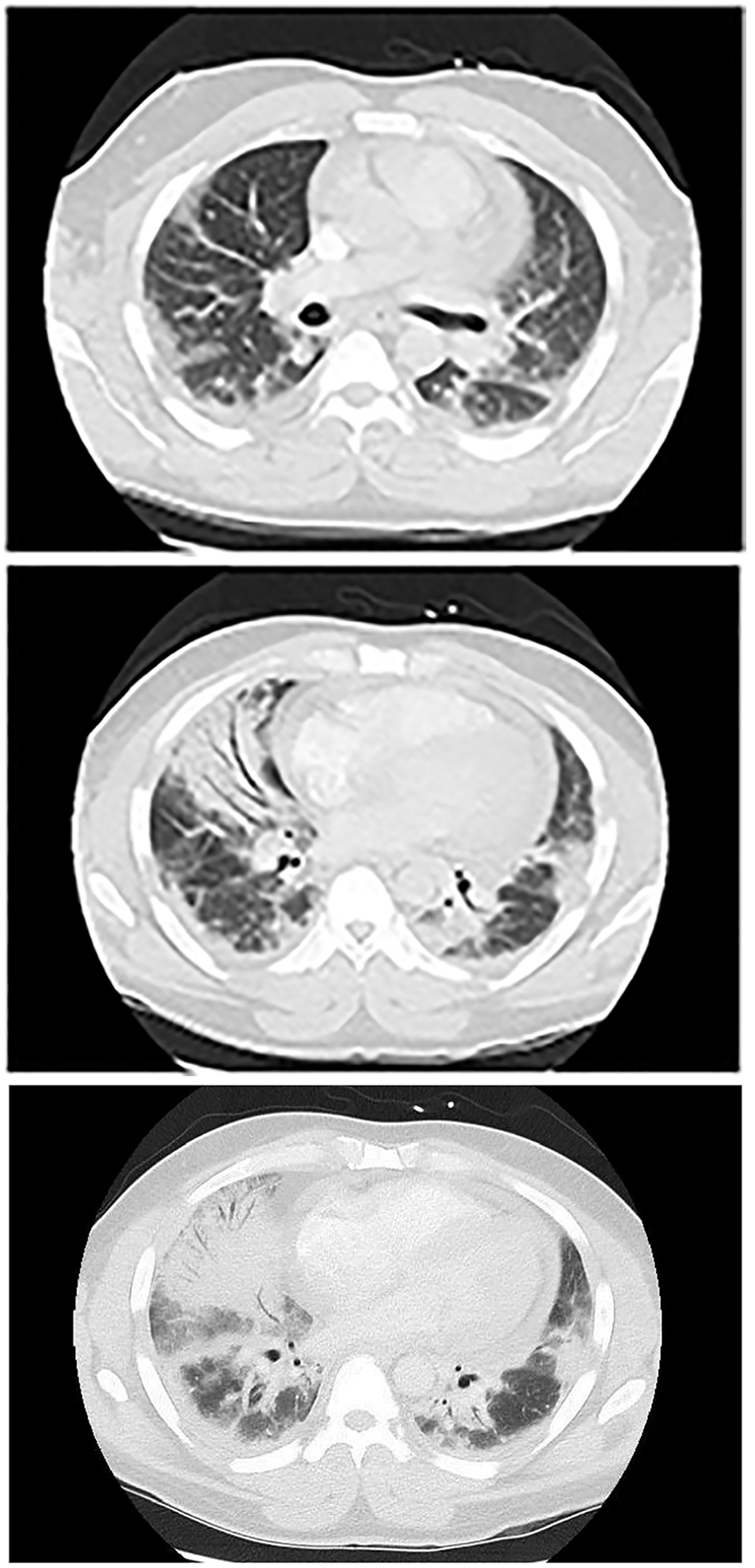 Click for large image | Figure 1. Pericardial effusion measuring 1.3 cm in thickness. Non-confluent areas of ill defined opacities were also seen in both lungs with denser consolidations and air Broncho-grams mainly in the right middle lobe and both the lower lobes which were thought to be a combination of infiltrates and atelectasis. |
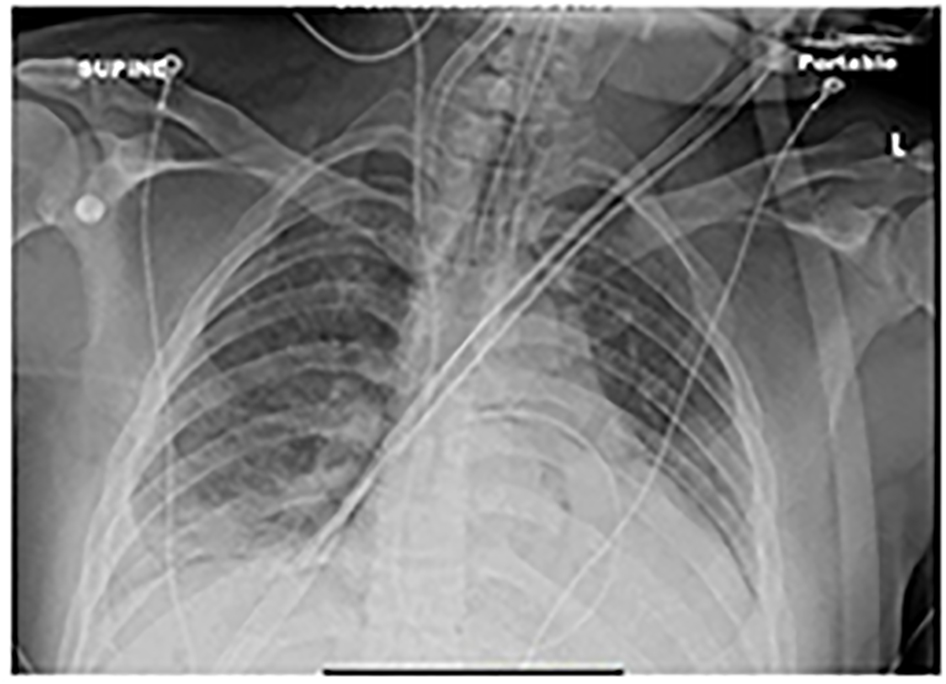 Click for large image | Figure 2. Small right-sided pleural effusion with a patchy right-sided opacity. |
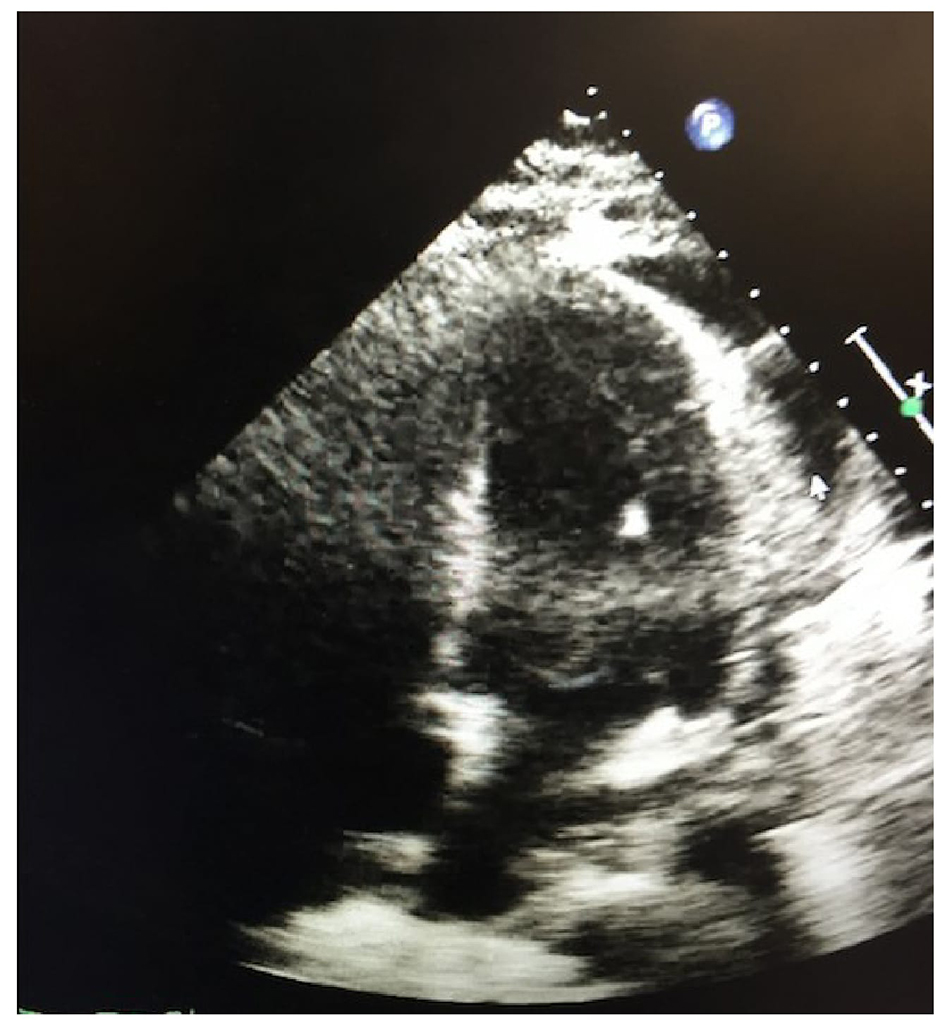 Click for large image | Figure 3. Echocardiogram (ECHO) evaluation revealed a moderate to large pericardial effusion. |
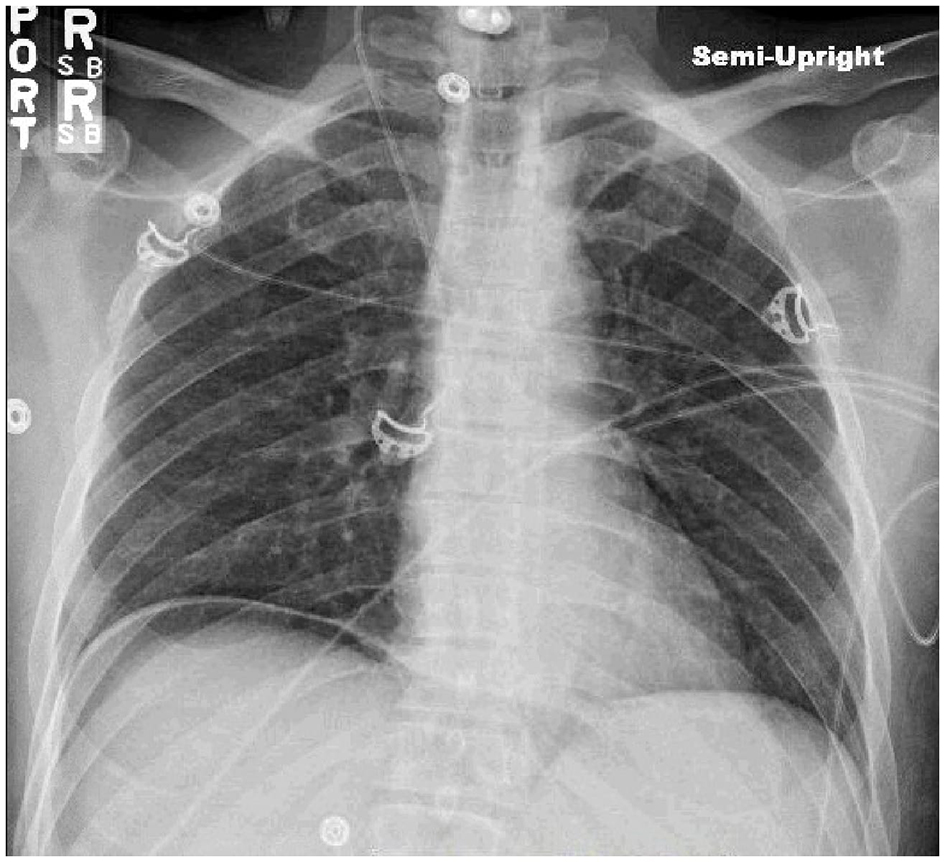 Click for large image | Figure 4. Complete resolution of the prior findings. |
The patient required intensive care unit transfer 2 days into admission for worsening hypoxia and hypotension initially treated with 24 h of non-invasive positive pressure ventilation and subsequent mechanical ventilation. Despite maximal ventilator support and pulmonary artery catheterization for optimal fluid management, the patient continued to decline. A decision to pursue veno-venous (VV) extracorporeal membrane oxygenation (ECMO) was undertaken for a diagnosis of severe acute respiratory distress syndrome 4 days into admission.
Owing to the radiographic and clinical findings suspicious for acute interstitial pneumonia and diagnostically negative bronchoscopic, viral, fungal, bacterial and mycobacterial serology, the patient was treated with broad spectrum antibiotics, namely, vancomycin, zosyn and azithromycin. The radiographic appearance, serologic findings combined with physical exam findings like “mechanic’s hands”, non-specific interstitial pneumonia with a pattern of ILD with elevated CK and positive anti-Jo-1, heightened the suspicion of a rare form of acutely progressing AS. Hence, the decision was made to treat the patient with high dose methylprednisolone 1 g daily for 3 days, followed by 125 mg, four times a day for 4 days, followed by a taper of 60 mg, four times a day for 4 days. The steroid medication was further weaned down to prednisone 40 mg daily, a dosage which he was discharged on.
With regard to his cardiovascular status, the patient had minimal vasopressor requirements despite his initial shock physiology. Follow-up serial echocardiography noted worsening pericardial effusion, necessitating the need for both diagnostic and therapeutic pericardial window. Fluid analysis showed no signs of infection with discharge surveillance ECHO showing no re-accumulation of pericardial fluid.
The patient developed non-oliguric renal failure thought to be secondary to acute tubular necrosis from a combination of hypotension and sepsis. The patient did not require renal replacement therapy however. Rhabdomyolysis was not felt to contribute to renal failure with CPKs peaking at 7,500 International Units/L followed by a gradual fall.
The course was also complicated by an upper gastrointestinal bleed which endoscopically was attributed to erosive gastritis managed conservatively via proton pump inhibitors.
Over the course of his stay, the patient showed significant improvement in his respiratory, cardiovascular and renal status and was decannulated from ECMO in 2 days (yes sure it was 2 days) with transition to mechanical ventilation. The patient was extubated shortly afterwards and had significantly improved clinical dyspnea.
| Discussion | ▴Top |
AS is a clinical entity which is described classically by the triad of ILD, inflammatory myositis and presence of ASA [1, 2]. Classical findings include “mechanics hands”, Raynaud’s phenomenon and polyarthritis [3]. The spectrum, presentation and course of this rare condition vary greatly but anti-Jo-1 antibody is the most commonly reported antibody [1, 2]. Pulmonary manifestations of the syndrome are associated with this antibody in most cases. Other possible antibodies associated with this syndrome are anti-PL-7, anti-PL-12, anti-EJ, anti-OJ, anti-KS, anti-Zo, anti-Ha-YRS, and anti-SRP [6].
There is no clear consensus for diagnosis of this syndrome except the demonstration of anti-ARS antibodies in the serum and ILD, myositis and arthritis. Presence of one of the above is considered sufficient for diagnosis [7]. Minor criteria for the diagnosis of AS include the presence of “mechanic’s hands”, Raynaud’s phenomenon and fever.
Anti-ARS antibodies are seen in 30-40% of patients with myositis [8], and in 7-10% of patients with idiopathic interstitial pneumonia [9]. The most common anti-ARS antibody is the anti-Jo-1 antibody (60% cases of AS), followed by anti-PL-7 and anti-PL-12 antibodies (10-15% cases of AS). Other antibodies are less common and are seen in less than 5% of AS patients [10]. Patients with anti-Jo-1 antibodies often present with arthritis, myositis and mechanics hands, whereas those with non-anti-Jo-1 antibodies present with fever and ILD [11].
Pulmonary involvement is encountered in majority of the cases of AS and is associated with morbidity as well as mortality [12]. The most common pulmonary manifestation of AS is the presence of ILD. The prevalence of ILD in patients with AS depends on the type of anti-ARS antibody (anti-Jo-1 vs. non-anti-Jo-1), clinicoradiological vs. radiological presentation; chest radiograph vs. HRCT chest, and the duration of follow-up [13]. The presence of anti-AS antibody is a stronger marker for ILD than myositis [14, 15]. For our patient, the clinical picture along with patchy consolidation and ground glass opacities in the lungs suggestive of non-specific interstitial pneumonia, supplemented by very high CRP, ANA titer of 1:320 and anti-Jo-1 antibody positive and elevated CK, raised the suspicion for AS, despite the uncharacteristically acute presentation of this condition. Though AS usually presents in a chronic fashion with gradually progressive muscle weakness and myopathy, the classic clinical, radiological and lab findings made us consider this in our differential. Of course the definitive diagnosis could have been strengthened by lung biopsy; however, owing to the clinical instability of the patient, this was not a feasible option and likely would not have changed management.
There are very few cases of AS occurring in such an acute fashion [16]. Our literature review found very few cases of this condition associated with pericardial effusion [17] and none necessitated the use of VV ECMO in the treatment of AS when examining the literature.
Unfortunately, treatment for the AS is limited, with use of immunosuppressive medications like glucocorticoids forming the basis of therapy. Serious complications of this condition include pulmonary hypertension and fibrosis. Prognosis is largely determined by the extent of pulmonary involvement and its progression.
Our patient had a rapidly progressing and deteriorating respiratory status requiring mechanical ventilation failure requiring ECMO. However, initiation of pulse dose steroids while bridging with VV ECMO improved the clinical course rapidly and the patient did well clinically and was discharged home with physical therapy assistance. He was instructed to wean off the steroid in a taper regimen as instructed by rheumatology with instructions to follow-up with them on an outpatient basis.
Conclusion
It would be prudent for clinicians, to keep this rare diagnosis in the differential of a patient with acute onset dyspnea, who has classic radiological and lab findings, along with a clinical picture suspicious for AS. Our case is rare with regard to the uncharacteristically acute nature of presentation, acute decompensation in clinical condition, development of acute interstitial pneumonitis requiring rescue ECMO and accompaniment of significant pericardial effusion on presentation, followed by rapid improvement with initiation of steroids.
| References | ▴Top |
- Bergoin C, Bure M, Tavernier JY, Lamblin C, Maurage CA, Remy-Jardin M, Wallaert B. [The anti-synthetase syndrome]. Rev Mal Respir. 2002;19(3):371-374.
pubmed - Hervier B, Wallaert B, Hachulla E, Adoue D, Lauque D, Audrain M, Camara B, et al. Clinical manifestations of anti-synthetase syndrome positive for anti-alanyl-tRNA synthetase (anti-PL12) antibodies: a retrospective study of 17 cases. Rheumatology (Oxford). 2010;49(5):972-976.
doi pubmed - Christopher-Stine L, Robinson DR, Wu CC, Mark EJ. Case records of the Massachusetts General Hospital. Case 37-2012. A 21-year-old man with fevers, arthralgias, and pulmonary infiltrates. N Engl J Med. 2012;367(22):2134-2146.
doi pubmed - Chatterjee S, Prayson R, Farver C. Antisynthetase syndrome: not just an inflammatory myopathy. Cleve Clin J Med. 2013;80(10):655-666.
doi pubmed - American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646-664.
pubmed - Lazarou IN, Guerne PA. Classification, diagnosis, and management of idiopathic inflammatory myopathies. J Rheumatol. 2013;40(5):550-564.
doi pubmed - Labirua-Iturburu A, Trallero Araguas E, Selva O'Callaghan A. [Anti-synthetase syndrome]. Med Clin (Barc). 2011;137(2):77-83.
doi pubmed - Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senecal JL. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther. 2007;9(4):R78.
doi pubmed - Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344-347.
doi pubmed - Aggarwal R, Cassidy E, Fertig N, Koontz DC, Lucas M, Ascherman DP, Oddis CV. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis. 2014;73(1):227-232.
doi pubmed - Lega JC, Fabien N, Reynaud Q, Durieu I, Durupt S, Dutertre M, Cordier JF, et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: a meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun Rev. 2014;13(9):883-891.
doi pubmed - Stanciu R, Guiguet M, Musset L, Touitou D, Beigelman C, Rigolet A, Costedoat-Chalumeau N, et al. Antisynthetase syndrome with anti-Jo1 antibodies in 48 patients: pulmonary involvement predicts disease-modifying antirheumatic drug use. J Rheumatol. 2012;39(9):1835-1839.
doi pubmed - Imbert-Masseau A, Hamidou M, Agard C, Grolleau JY, Cherin P. Antisynthetase syndrome. Joint Bone Spine. 2003;70(3):161-168.
doi - Matsushita T, Hasegawa M, Fujimoto M, Hamaguchi Y, Komura K, Hirano T, Horikawa M, et al. Clinical evaluation of anti-aminoacyl tRNA synthetase antibodies in Japanese patients with dermatomyositis. J Rheumatol. 2007;34(5):1012-1018.
pubmed - Schmidt WA, Wetzel W, Friedlander R, Lange R, Sorensen HF, Lichey HJ, Genth E, et al. Clinical and serological aspects of patients with anti-Jo-1 antibodies - an evolving spectrum of disease manifestations. Clin Rheumatol. 2000;19(5):371-377.
doi pubmed - Jordan Greco AS, Metrailler JC, Dayer E. [The antisynthetase syndrome: a cause of rapidly progressive interstitial lung disease]. Rev Med Suisse. 2007;3(134):2675-2676, 2679-2681.
pubmed - Maturu VN, Lakshman A, Bal A, Dhir V, Sharma A, Garg M, Saikia B, et al. Antisynthetase syndrome: An under-recognized cause of interstitial lung disease. Lung India. 2016;33(1):20-26.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.