

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Original Article
Volume 8, Number 5, May 2016, pages 379-384
Recombinase Activating Gene 1 Deficiencies Without Omenn Syndrome May Also Present With Eosinophilia and Bone Marrow Fibrosis
Ezgi Ulusoya, b, Neslihan Edeer Karacaa, Elif Azarsiza, Afig Berdelia, Guzide Aksua, Necil Kutukculera
aDepartment of Pediatrics, Faculty of Medicine, Ege University, Izmir, Turkey
bCorresponding Author: Ezgi Ulusoy, Department of Pediatrics, Faculty of Medicine, Ege University, 35100 Bornova, Izmir, Turkey
Manuscript accepted for publication September 11, 2015
Short title: RAG1 Deficiencies
doi: http://dx.doi.org/10.14740/jocmr2316w
| Abstract | ▴Top |
Background: Severe combined immunodeficiency (SCID) syndromes are a heterogenous group of diseases characterized by impairment in both cellular and humoral immunity with a range of genetic disorders. Complete recombinase activating gene (RAG) deficiency is associated with classical T-B-NK+ SCID which is the most common phenotype of Turkish SCID patients. There is a broad spectrum of hypomorfic RAG mutations including Omenn syndrome, leaky or atypical SCID with expansion of γδ T cells, autoimmunity and cytomegalovirus (CMV) infections.
Methods: Twenty-one (44%) patients had RAG1 deficiency of all 44 SCID patients followed up by pediatric immunology department. A retrospective analysis was conducted on the medical records of all SCID patients with RAG1 deficiency.
Results: Eight patients were classified as T-B-NK+ SCID, five patients as T+B-NK+ SCID (three of these were Omenn phenotype), and eight patients as T+B+NK+ SCID phenotype. Mean age of the whole study group, mean age at onset of symptoms and mean age at diagnosis were 87.7 ± 73.8 (12 - 256), 4.4 ± 8.2 (1 - 36) and 29.1 ± 56.8 (1 - 244) months, respectively. Consanguinity was present in 11 (52%) of 21 patients. Autoimmunity was found in six patients (28%). Ten patients (47%) had CMV infection, four (19%) had Epstein-Barr virus (EBV) infections and three (14%) had Bacillus Calmette-Guerin (BCG) infections. Seven patients who had refractory cytopenia (two pancytopenia and five bicytopenia) underwent bone marrow biopsy, three of whom had bone marrow fibrosis. Future evaluations must be considered about bone marrow fibrosis in RAG1 deficiency patients. Eosinophilia was observed in 10 patients, seven of whom did not have Omenn phenotype.
Conclusion: Non-Omenn phenotype RAG1 deficiencies can also present with eosinophilia. This report is presented to emphasize that RAG1 mutations may lead to diverse clinical phenotypes.
Keywords: Eosinophilia; Fibrosis; Recombinase activating gene
| Introduction | ▴Top |
Severe combined immunodeficiency (SCID) syndromes are a heterogenous group of diseases characterized by impairment in both cellular and humoral immunity with a range of genetic disorders. SCID patients present with recurrent life threatening infections in the first year of life and the only curative treatment is hematopoetic stem cell transplantation (HSCT) [1-3].
Recombinase activating genes 1 and 2 (RAG1 and RAG2) located in chromosome 11p13 are essential for the process of rearrangement of the variable (V), diversity (D) and joining (J) segments during T- and B-cell development [1, 4-6]. The adaptive immune system forms millions of antigen-specific receptors by the process of V(D)J recombination. Complete RAG deficiency (RAGD) without a V(D)J rearrangement is associated with classical T-B-NK+ SCID. T-B-NK+ SCID was reported to be the most common SCID type in Turkey (28%) [7]. In Kilic et al’s study, SCID prevalence was 2% of all primary immunodeficiency diseases. RAG1 and two genes are responsible for 50% of all patients with this SCID phenotype [8-10]. Defects in Artemis, DNA-ligase IV, Cernunnos and DNA-PKcs also cause T-B-NK+ SCID [11-14].
Schwarz et al [15] first described human autosomal recessive SCID patients with non-sense, missense and deletional mutations in the RAG1 and RAG2 genes in 1996. Further evaluation showed a broad spectrum of hypomorphic RAG mutations including Omenn syndrome (with skin inflammation and αβ T-cell expansion), leaky or atypical SCID form with expansion of γδ T cells, autoimmunity and cytomegalovirus (CMV) infections and RAGD characterized by granulomatous lesions in skin and other tissues [3, 16-18].
The aim of this study was to review the clinical, immunological and molecular findings of Turkish SCID patients with RAG1 defects and to draw attention to the bone marrow fibrosis in some patients and high frequency of eosinophilia even in non-Omenn phenotype.
| Patients and Methods | ▴Top |
A retrospective analysis was conducted on the medical records of all SCID patients with RAG1 deficiency, diagnosed at Ege University, Faculty of Medicine, Department of Pediatric Immunology between 2002 and 2015.
Twenty-one SCID patients with RAG1 deficiency of all 44 SCID patients were included in this study. Data on demographic information of patients including name, gender, date of birth, age of onset of symptoms, clinical symptoms, age at diagnosis, family history and consanguinity, previous history of medications and vaccination, laboratory and molecular data were collected retrospectively by an inquiry form. Autoimmunity, lymphoproliferation, chronic lung disease, blood marrow fibrosis, eosinophilia, CMV, Epstein-Barr virus (EBV) and Bacillus Calmette-Guerin (BCG) infections were all investigated.
Complete blood count with peripheral blood smear evaluation, serum immunoglobulins by nephelometry (Dade Behring BNII Nephelometer Analyzer, Germany) and lymphocyte phenotyping (T, B, and NK cells) by flow cytometry (FACSCalibur, Becton-Dickinson, USA) were the standard laboratory investigations performed for our patients. On the basis of phenotype, each patient was assigned to one of the following subgroups: T-B-NK+ SCID, Omenn syndrome, T+B-NK+ and T+B+NK+ phenotype.
Mutation analyses of RAG1 gene were performed as previously described [4]. Data analyses were performed using Statistical Package for Social Sciences version 16.0.
| Results | ▴Top |
Twenty-one patients (F/M: 10/11) with RAG1 deficiency were included. Their demographic findings are listed in Table 1. Eight patients (38%) were classified as T-B-NK+ SCID, five patients (23%) as T+B-NK+ SCID (three of these patients were Omenn syndrome), eight patients (38%) as T+B+NK+ SCID (leaky SCID) phenotype (Fig. 1).
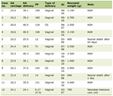 Click to view | Table 1. Demographic and Clinical Findings of Patients With RAG1 Deficiency |
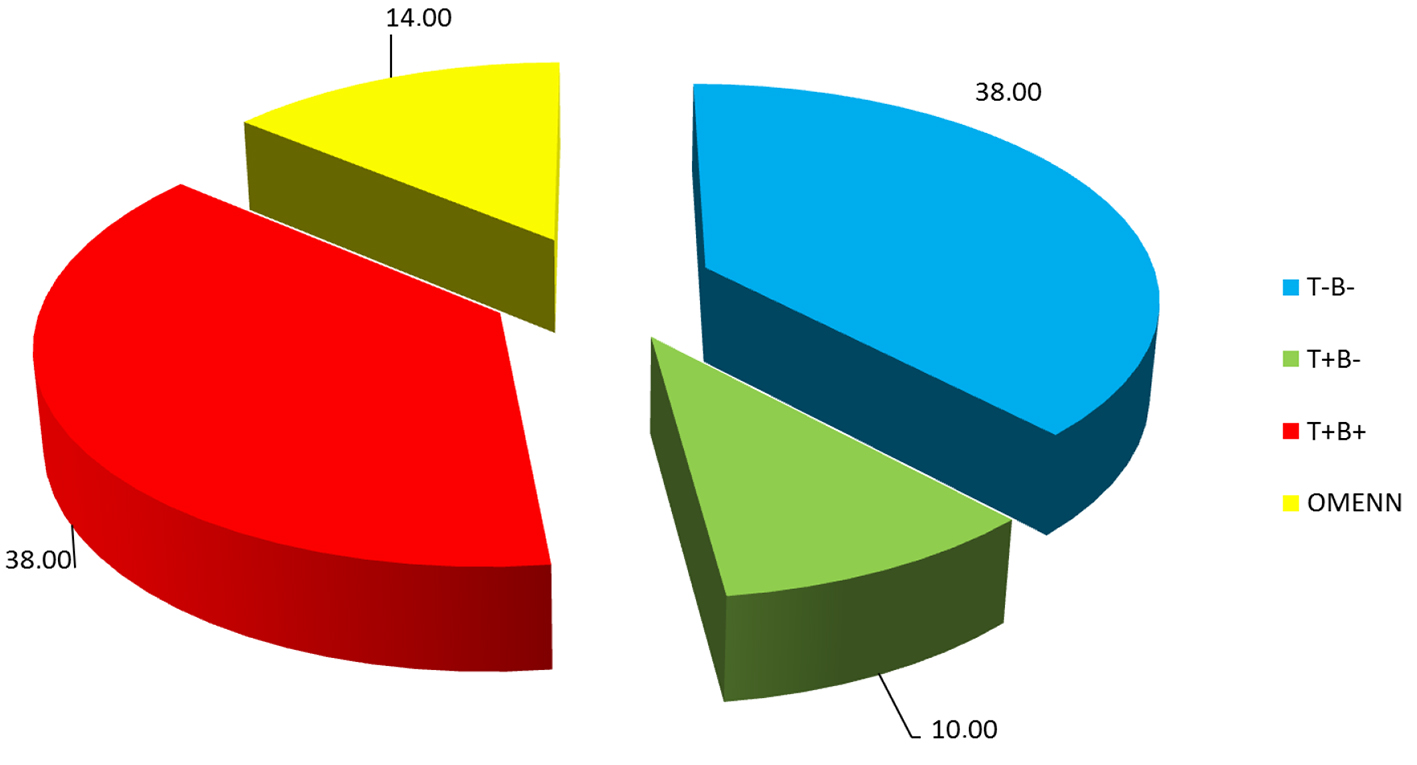 Click for large image | Figure 1. Distribution of patients with RAG1 deficiency. |
Mean age of the whole study group, mean age at onset of symptoms and mean age at diagnosis were 87.7 ± 73.8 (12 - 256), 4.4 ± 8.2 (1 - 36) and 29.1 ± 56.8 (1 - 244) months, respectively (Table 1). Consanguinity rate was 11 (52%) in 21 patients. Mean age at onset of symptoms and age at diagnosis were statistically older in patients with T+B+ group (P = 0.005 and P = 0.001, respectively).
Autoimmune findings were observed in six patients (28%) (two had autoimmune hemolytic anemia and four had positive Coombs test with no clinical signs). Ten patients (47%) had CMV infection, four (19%) had EBV infections, and three (14%) had BCG infections (two with BCGitis and one with BCGosis) (Table 1). Twelve of all patients (57%) had failure to thrive. Organomegaly was found in 11 (52%) patients. Ten patients were complicated with chronic lung disease. All three patients with Omenn phenotype presented with erythroderma and eosinophilia. Also one patient in T+B+NK+ SCID phenotype had erythroderma. In this group, seven patients underwent bone marrow biopsy for different indications (two pancytopenia and five bicytopenia) and three of them had bone marrow fibrosis (Table 2).
Click to view | Table 2. Clinical Findings of Patients With RAG1 Deficiency |
Lymphopenia was documented in six of all patients (28%). Five patients had neutropenia and four patients had thrombocytopenia. Eosinophilia was determined in 10 patients, seven of whom were not Omenn syndrome (Table 3). None of these patients had any parasitic infections or any other pathologies explaining eosinophilia.
Click to view | Table 3. Hematologic Evaluation of Patients With RAG1 Deficiency |
Lymphocyte subset distribution and increase in HLA-DR, TCRγδ+ expression on T cells are shown in Table 4. HLA-DR elevation was determined in four patients (80%) with T+B-NK+ SCID phenotype including Omenn phenotype and in eight (38%) of all patients. γδ elevation was documented in four patients, one in T+B-NK+ SCID and three in T+B+NK+ SCID phenotype.
Click to view | Table 4. Lymphocyte Subsets of Patients With RAG 1 Deficiency |
All patients with RAG1 mutations are shown in Table 5.
Click to view | Table 5. RAG1 Mutations and Clinical Outcomes of Patients |
In this group, nine patients underwent HSCT, four from unrelated donors. After HSCT, two patients died, and the other seven patients recovered. Six patients who did not have related donors died during the follow-up period before an unrelated donor was found (Table 5).
| Discussion | ▴Top |
RAG1 and RAG2 deficiencies are responsible for 10% of all SCID cases. Phenotypic distrubition of this group was 38% T-B-NK+ SCID, 23% T+B-NK+ SCID and 38% T+B+NK+ SCID (leaky SCID) phenotype.
The relative frequency of the different forms of SCID may vary in different countries because of genetic, social and environmental differences. Among Turkish patients, the most common phenoype is T-B-NK+ SCID. RAG1 deficiency is inherited with an autosomal recessive pattern. In this group, consanguinity rate was high (52%). Risk of autosomal recesive disorders is high because of high consanguineous marriages in Turkish population.
Mean age at onset of symptoms was 4.4 ± 8.2 months, but mean age at diagnosis was 29 ± 56 months. This difference between onset of the symptoms and diagnosis is especially because of the clinical heterogenity and late diagnosis of leaky SCID phenotype.
SCID has a very wide spectrum including late onset combined immunodeficiency (CID) or leaky SCID with hypomorphic RAG mutations [19]. Villartay et al [16] first described four patients from four different families. These patients had severe CMV infection with TCRγδ expansion and autoimmunity. In our group, not only leaky SCID patients but also three of the T-B-NK+ patients and one patient with Omenn phenotype had CMV infection. One patient with T+B-NK+ SCID (non-Omenn phenotype) had CMV infection with HLA-DR and γδ elevation. This patient had the same homozygous mutation H561R with the patient discribed by Ehl et al previously with predominance of γδ T cells and CMV infection [3]. One of the patients in T+B+NK+ SCID group had the same mutation (del AA368-369) with one of Villartay’s patients and he also had CMV infection with TCR γδ expansion and autoimmunity. Four of our patients also had EBV infections.
SCID patients can also present with BCG infections [20, 21]. Two of our patients with T-B-NK+ phenotype had BCGitis and one patient with leaky SCID phenotype had skin BCG abscess.
All patients with T+B+NK+ SCID phenotype had chronic lung diseases. The percentage was more than the other phenotypes. This fact can be associated with the older age of this group and as a result more exposure to infections.
Some patients with RAGD may present later in life as CID with granuloma formation, none of our patients, including patients with late onset, had granuloma formation so far [17].
One of the most remarkable parts of this study is the high percentage of eosinophilia observed in non-Omenn patients (33%). Eosinophilia was present in 67.5% of T-B-NK+ SCID patients. Parasitic infections and the other pathologies, such as allergic reactions, Hodgkin lymphoma or myeloproliferative disorders causing eosinophilia were excluded. There was not any significant correlation between eosinophilia and type of mutations. This suggests us that non-Omenn phenotype SCID patients with RAG1 deficiency can also present with eosinophilia. Eosinophilia is also reported in primary immunodeficiency patients other than RAG defects, such as congenital neutropenias, adenosine deaminase and DOCK8 deficiencies (literaür eklenecek). Compensatory mechanisms may be the reason for the increase in eosinophil counts in these patients. Further evaluation is needed about eosinophilia in these patients.
SCID patients mostly have lymphopenia. In the follow-up period, some patients can develop other cytopenias such as thrombocytopenia, and neutropenia which can persist for a long time. In our group, seven patients underwent bone marrow biopsies because of persistent cytopenia and bone marrow fibrosis was determined in three of these patients. To our knowledge, there are no reports about bone marrow fibrosis in RAG deficient patients. Two of these three patients had the same compound heterozygous mutation H249R/K820R and one of them also had BCG infection.
Different mutations may lead to different phenotypes and the same mutation may present with different phenotypes. In our group, H249R and K820R compound heterozygous and H249R homozygous or heterozygous mutations were predominant. H249R/K820R compound heterozygous mutations were found in all SCID phenoypes. The H249R heterozygous mutation was also fond in all phenotypes except T+B-NK+ phenotype. This also suggests us that same mutations can present with different phenotypes. These mutations were reported as polymorphisms previously [22]. Two patients having H249R/K820R compound heterozygous mutations were cousins with the same phenotype of T-B-NK+ SCID. Investigating RAG activity in our patients will give us more accurate information about these defects being the reason of clinical phenotypes of our patients. We also describe a novel mutation p. Leu846Pr and the patient had classic T-B-NK+ SCID phenotype.
We wanted to pay attention to eosinophilia of RAG1 deficient patients with non-Omenn phenotype. We also detected three patients with bone marrow fibrosis in our group and this can be a clinical part of RAG1 deficiencies. Further evaluations must be considered.
| References | ▴Top |
- Dhingra N, Yadav SP, de Villartay JP, Picard C, Sabharwal RK, Dinand V, Ghuman SS, et al. Severe combined immunodeficiency caused by a new homozygous RAG1 mutation with progressive encephalopathy. Hematol Oncol Stem Cell Ther. 2014;7(1):44-49.
doi pubmed - Gaspar HB. Tearing RAGs apart. Blood. 2014;123(2):156-157.
doi pubmed - Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kuhr J, Mascart F, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest. 2005;115(11):3140-3148.
doi pubmed - Kutukculer N, Gulez N, Karaca NE, Aksu G, Berdeli A. Novel mutations and diverse clinical phenotypes in recombinase-activating gene 1 deficiency. Ital J Pediatr. 2012;38:8.
doi pubmed - Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, Arkwright PD, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood. 2001;97(1):81-88.
doi pubmed - Jones JM, Simkus C. The roles of the RAG1 and RAG2 "non-core" regions in V(D)J recombination and lymphocyte development. Arch Immunol Ther Exp (Warsz). 2009;57(2):105-116.
doi pubmed - Kilic SS, Ozel M, Hafizoglu D, Karaca NE, Aksu G, Kutukculer N. The prevalences [correction] and patient characteristics of primary immunodeficiency diseases in Turkey - two centers study. J Clin Immunol. 2013;33(1):74-83.
doi pubmed - Kato T, Crestani E, Kamae C, Honma K, Yokosuka T, Ikegawa T, Nishida N, et al. RAG1 deficiency may present clinically as selective IgA deficiency. J Clin Immunol. 2015;35(3):280-288.
doi pubmed - Niehues T, Perez-Becker R, Schuetz C. More than just SCID - the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2. Clin Immunol. 2010;135(2):183-192.
doi pubmed - Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625-655.
doi pubmed - Lee PP, Woodbine L, Gilmour KC, Bibi S, Cale CM, Amrolia PJ, Veys PA, et al. The many faces of Artemis-deficient combined immunodeficiency - Two patients with DCLRE1C mutations and a systematic literature review of genotype-phenotype correlation. Clin Immunol. 2013;149(3):464-474.
doi pubmed - Enders A, Fisch P, Schwarz K, Duffner U, Pannicke U, Nikolopoulos E, Peters A, et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J Immunol. 2006;176(8):5060-5068.
doi pubmed - Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162.
pubmed - Bousfiha AA, Jeddane L, Ailal F, Al Herz W, Conley ME, Cunningham-Rundles C, Etzioni A, et al. A phenotypic approach for IUIS PID classification and diagnosis: guidelines for clinicians at the bedside. J Clin Immunol. 2013;33(6):1078-1087.
doi pubmed - Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, Friedrich W, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274(5284):97-99.
doi pubmed - de Villartay JP, Lim A, Al-Mousa H, Dupont S, Dechanet-Merville J, Coumau-Gatbois E, Gougeon ML, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest. 2005;115(11):3291-3299.
doi pubmed - Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, Schneider DT, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008;358(19):2030-2038.
doi pubmed - De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, Douek DC, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood. 2010;116(8):1263-1271.
doi pubmed - Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, Nugent D, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol. 2015;35(2):119-124.
doi pubmed - Sadeghi-Shanbestari M, Ansarin K, Maljaei SH, Rafeey M, Pezeshki Z, Kousha A, Baradaran R, et al. Immunologic aspects of patients with disseminated bacille Calmette-Guerin disease in north-west of Iran. Ital J Pediatr. 2009;35:42.
doi - Mittal H, Faridi M, Kumar P, Aggarwal A. Disseminated bacillus calmette guerin disease in a twin infant with severe combined immunodeficiency disease. J Glob Infect Dis. 2014;6(3):132-134.
doi pubmed - Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, Al-Herz W, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. 2014;133(4):1099-1108.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.