

| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Original Article
Volume 1, Number 4, October 2009, pages 212-218
Studies on Genomic DNA Stability in Aluminium-Maltolate Treated Aged New Zealand Rabbit: Relevance to the Alzheimers Animal Model
Obulesu Magisettya, Dowlathabad Muralidhara Raob, c, Shama Sundar N. Ma
aDepartment of Anatomy, JSS Medical College, Mysore, India
bDepartment of Biotechnology, Sri Krishnadevaraya University, Anantapur, India
cCorresponding author:
Manuscript accepted for publication August 19, 2009
Short title: DNA Damage in AD
doi: https://doi.org/10.4021/jocmr2009.09.1265
| Abstract | ▴Top |
Background: Alzheimers disease (AD) is a devastative neurodegenerative disorder. Lack of substantial animal model that can unravel molecular underpinnings has been a major lacuna which limited the understanding of the etiology of the disease in turn limiting the employment of potential therapeutic strategies to combat the disease for a few decades. Our studies for the first time provided substantial animal model and tattered the etiology of the disease at a molecular level.
Methods: In this study DNA was isolated from Hippocampus (H), Midbrain (M) and Frontal Cortex (Fc) of control and aluminium maltolate (Al-M) treated aged New Zealand rabbit brain. DNA damage has been studied using Agarose gel electrophoresis, Ethidium Bromide (EtBr) binding and Melting temperature techniques.
Results: Al-M treated aged New Zealand rabbit's H and M showed higher DNA damage compared to corresponding controls, where as Fc showed mild DNA damage compared to corresponding controls.
Conclusions: This study tangibly provides substantial molecular level understanding of the disease in turn providing an adequate platform to streamline potential therapeutic strategies.
Keywords: Alzheimer’s disease; Aluminium maltolate; Animal model; DNA damage
| Introduction | ▴Top |
Although adequate research has been progressing for a century, the understanding of the etiology of Alzheimers disease (AD) has not been completely unraveled due to the lack of substantial animal model [1,2]. The biochemical events entailed in the neuronal cell loss are not clear till date [1,3]. The majority of the animal models worked till date could demonstrate single event expression such as extra cellular Aβ deposition, intraneuronal neurofilamentous aggregation of proteins similar to neurofibrillary tangles, oxidative stress and apoptosis [1,4]. However, the intracisternal injection of aluminium maltolate (Al-M) into aged New Zealand rabbit replicates the neuropathological, biochemical and behavioural changes found in AD [1, 5-13]. Lovell et al reported DNA damage like single strand/double strand breaks, base specific oxidation like G* specific oxidation [14-19]. Increased DNA oxidation has also been noticed in AD [20-22]. A few studies have shown that DNA repair failure is a prominent feature in AD [23,24]. The deterioration of the DNA repair system takes place with aging. Abnormal cell cycle regulation and/or accumulated DNA damage also leads to AD [25]. A few animal models were developed to study the genes responsible for this disease and the increase of specific transcripts [26]. The pathological events like hyperphosphorylation of tau, formation of neurofibrillary tangles, Aβ deposition, were studied [27-29]. Introduction of Aβ 1-42 into rabbits also provided animal model for AD [30].
Accumulation of DNA damage may also lead to AD [31]. Limited work has been done on DNA stability in this animal model so far. The present work has been carried out to unravel the molecular underpinnings of the disease pathology.
| Materials and Methods | ▴Top |
Maltol, Al (NO3)3. 9 H2O, Agarose, Ethidium Bromide (EtBr), Hepes, Tris, Hepes and Tris buffers were purchased from Sigma Chemicals (USA). All other chemicals were of analytical grade and were purchased from Sisco Research Labs, Mumbai, India.
Al-M preparation
Al-M was prepared from maltol (3-hydroxy-2methyl-4H pyran-4-one) following the method of Finneagan [32]. Maltol and Al (NO3)3. 9 H2O were mixed in 3:1 ratio. The pH was adjusted to 8.6 and heated for a few minutes. This aluminium-maltolate was used in the present study to understand the Aluminium's effect on genomic DNA. Aluminium complex is hydrolytically stable from pH 2.0 to 12.0. This complex enhances free Aluminium existence by 60-70% at neutral pH compared to any other inorganic or organic Aluminium complex.
Aluminium speciation chemistry is a complex phenomenon, hence to overcome this observation Aluminium-maltolate (Al-M) was used in the present investigation.
Animal treatment and tissue processing
All the animals were maintained in JSS animal house in single stainless steel cages. The experiments were done according to the institutes ethical committee and INSA guidelines. Aged Rabbits (3.5 to 4 yrs) from JSS Medical College Animal Colony were used. Six Rabbits were treated with Al-M intracisternally, while 6 rabbits with 0.9% saline injection were used as control. The intracisternal injection of Al-M into aged New Zealand rabbits was done as described by Savory [11,33]. The animals were decapitated and their brains were quickly removed, snap-frozen in N-methylbutane at a temperature of -40 oC with liquid nitrogen and stored at -80 oC [34]. The natomical localization of hippocampus, midbrain and cortical regions were accomplished using the designation outlined in an atlas of the rabbit brain and spinal cord [35].
Isolation of DNA from brain tissue
Genomic DNA from control and Al-M treated brain tissues were isolated by phenol-chloroform extraction protocol [36]. Tissue pieces were transferred into an autoclaved porcelain mortar and pestle (all glass wares, mortar, pestle were autoclaved before using them in order to avoid bacterial contamination). Liquid nitrogen was poured into the mortar and the tissue was frozen. Tissue was ground thoroughly with pestle with frequent additions of liquid nitrogen. Sufficient quantity of liquid nitrogen was poured into the mortar and swirled. Tissue homogenate was transferred into a sterile tube and the liquid nitrogen was allowed to evaporate. A sterile spatula was used to transfer the powdered tissue into a graduated tube. Lysis buffer 50 mM Tris-Hcl (pH 8.0), 10 mM EDTA, and 100 mM NaCl was added into the tube along with 15 ug per ml of proteinase K and 2% SDS final volume. One ml of lysis buffer was used for every 500 mg of tissue. Lysis buffer was pre warmed, added proteinase K after first 2h. The homogenate was incubated at 37 oC in a water bath for 12-16 h or over night. After the completion of incubation, the incubated lysate was transferred to an autoclaved 50 ml conical flask and equal volume of tris-saturated phenol (pH 8.0) was added and mixed thoroughly, either manually or mechanically for 10 min. The lysate was centrifuged for 10 min at 10,000 rpm at 13 oC. The supernatant was collected into a fresh autoclaved 50 ml conical flasks and half volume of tris saturated phenol and chloroform: isoamyl alcohol was added and mixed thoroughly. One part phenol: 1 part chloroform (C) and isoamylalcohol (IA) mixture (C: IA= 23:1). Tris-saturated phenol was stored in amber colored bottles at low temperature to avoid oxidation of phenol. The supernatant and tris-saturated phenol-chloroform mixture was centrifuged at 5000 rpm at 4 oC.
The upper aqueous layer was collected into a fresh tube and 1/3 volume of sodium acetate (pH 5.5) and equal volume of chilled absolute ethanol was added. DNA was precipitated by slowly swirling the tube manually. DNA was washed twice with 70% alcohol and once with absolute alcohol to remove excess salt and vacuum dried. The vacuum dried DNA was dissolved in 1 ml of TE buffer (10 mM Tris-Hcl 1mM EDTA, pH 8.0). The DNA isolated from cells contains RNA also which was removed by digesting the preparation with RNAse enzyme. RNAse solution was kept in boiling water for 10 min so as to inactivate any DNAse because the RNAse may contain DNAse also. RNAse can also be added before Proteinase K treatment, and incubated at 37 oC for 1 hour then start Proteinase K treatment (Add 1 g of RNAse/ml of lysis buffer for 30 min).
DNA concentration and purity
DNA was quantified by recording its optical density at 260 nm by UV spectrophotometer. Absorbance of isolated DNA was read at 280 nm. A260/A280 nm ratio was measured [36].
Neutral agarose gel electrophoresis
The genomic DNA integrity and damage was assessed by running neutral gel electrophoresis. The migration pattern in neutral gels reflects the double strand breaks present in the DNA. Neutral gels were electrophoresed on 1.5% agarose gels in Tris-acetate EDTA buffer (pH 8.0) at 4V/cm for 4h. Three ug of DNA was loaded in each well [36].
EtBr binding to DNA and Scatchard plots
The binding of EtBr to control and Al-M treated aged rabbits brain DNA was measured in 0.01 M HEPES buffer (pH 7.0) using Spectrofluorimeter (HITACHI, Japan) by taking 1:1 (w/w) DNA/EtBr before measuring fluorescence emission. DNA/EtBr solutions were excited at 535 nm, and emission intensity was monitored at 600 nm using HITACHI F-2000 Fluorescence Spectrophotometer. The amount of EtBr bound to DNA was calculated using the independent binding equation of Scatchard [36,37].
Melting temperature profiles
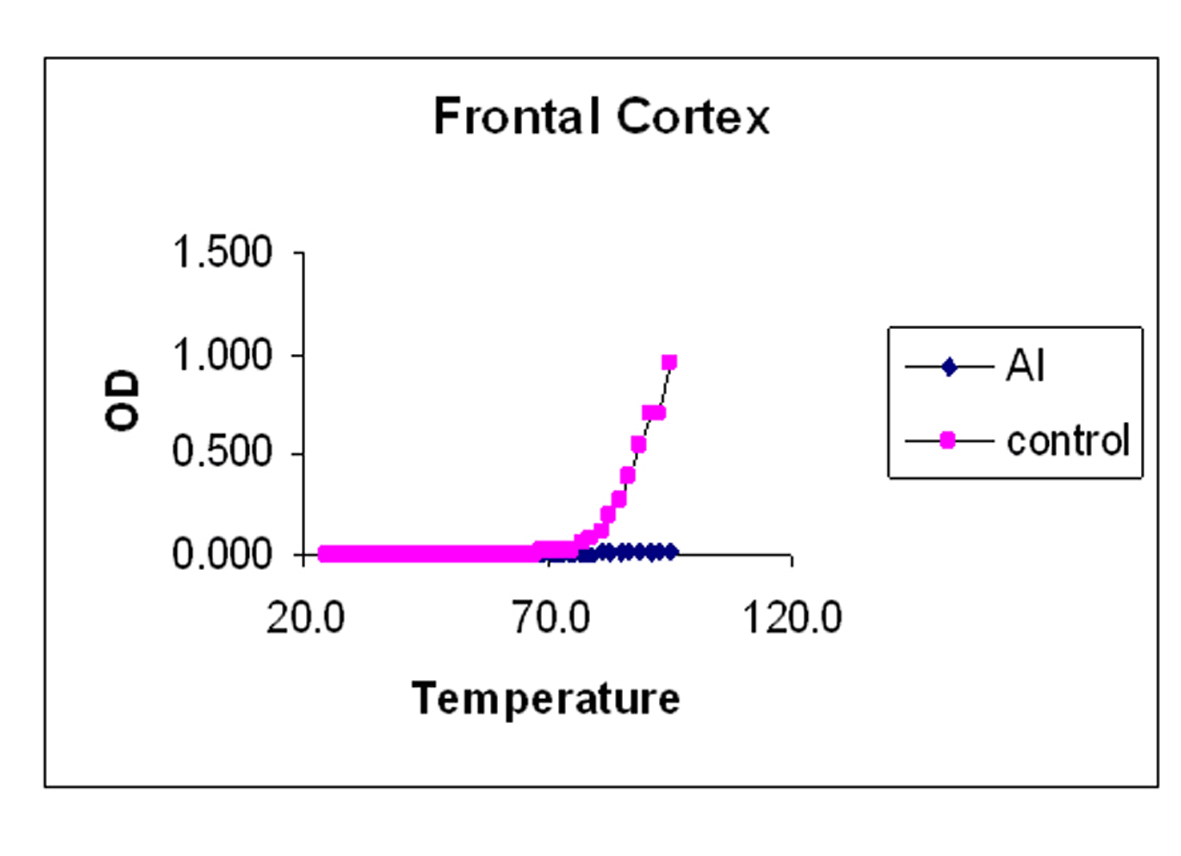
The melting curves for control and Al-M treated rabbits brain DNA was carried out in 0.01 M HEPES buffer (pH 7.4) by recording UV absorbance at 260 nm at different temperatures 1 oC /min using a Gilford Response II UV spectrophotometer fitted with thermostat control. The temperature range was between 25-95 oC. Tm values were determined graphically from the absorbance versus temperature plots. The temperature point at which there is a 50% absorbance shift was taken as melting temperature (Tm) of the DNA sample. The precision of Tm values was estimated [36].
| Results | ▴Top |
Behavioural features
Al-M treated aged New Zealand rabbits showed following behavioural changes like reduced food intake, isolation behavior, forward head tilting, increased eye blinking, and hemiplegia.
Agarose Gel electrophoresis
DNA damage was observed by neutral agarose gel electrophoresis. Figure 1 shows neutral gels of genomic DNA from H of control and Al-M treated aged New Zealand white rabbits. Figure 2 shows neutral gels of genomic DNA from M of control and Al-M treated aged New Zealand white rabbits. Figure 3 shows neutral gels of genomic DNA from Fc of control and Al-M treated aged New Zealand white rabbits.
 Click for large image | Figure 1. Indicates migration pattern of control and Al-M treated H DNA. Left lane M is Lambda DNA / EcoRI/HindIII double digest marker, 2-7 are control samples of H showing intact DNA where as 8-13 showed diffused bands indicating DNA damage. |
| Discussion | ▴Top |
Genomic DNA integrity plays pivotal role in the survival of an organism. The damage caused to its integrity shows deleterious effects on the health eventually leading to death. DNA repair capacity reduces with advancing age thus leading to the accumulation of the DNA damage. Oxidation of bases, strand breaks, formation of adducts, are the considerable factors in AD pathogenesis [15]. The animals tested so far could not provide substantial information on the etiology of the disease.
The animal models studied till date could demonstrate single pathological feature but failed to reciprocate the complete pathology of the AD. This lacuna made the insight into the pathology of the disease an uphill task for a few decades. Al is known to induce neurodegeneratiion although its role has been suspicious for a few decades. Since the advent of neurotoxicological studies many Al salts like AlCl3 [38] have been employed which produced insoluble complexes at neutral pH. Al-M has been chosen to be a neurotoxic agent for our studies, since maltolate aggravates the neurotoxicity of the Al. Al-M induced rabbits mimic AD pathology. Al-M has following properties which made it an appropriate neurotoxin: 1, very high metal solubility at pH 7.0; 2, prominent kinetic restrictions to ligand exchange reactions in neutral solution [1]. This work accentuates the role of Al in neurodegeneration.
Our studies showed DNA damage in Al-M treated aged rabbits. Gel pattern showed the damage which is substantiated by Tm and EtBr values. Low Tm and less binding of EtBr to the DNA emphasize the DNA damage [36].
Although adequate research has been going on for almost a century, the etiology of the AD has not yet been unraveled completely. The above observations emphasize the fact that there is severe damage in H which leads to the memory loss. The next highest damage has been noticed in M. The damage observed in Fc is considerably less. These findings are in accordance with the AD pathology. DNA damage or stability reduction plays pivotal role in AD. This work throws light on molecular understanding of pathology and tangibly gives substantial information about the etiology of the disease which stands as a suitable corner stone to streamline the potential therapeutic strategies.
Acknowledgments
This work is supported by Indian Council of Medical Research. M. Obulesu profoundly thanks Dr. KSJ Rao, Scientist, CFTRI, Mysore and Dr. K.H. Basavaraj for their support. He also sincerely thanks Indian Council of Medical Research for awarding Senior Research Fellowship.
| References | ▴Top |
- Bharathi, Shama Sundar NM, Rao TSS, Ravid R, Rao KSJ. A new insight into Al-maltolate treated aged rabbit brain as Alzheimer’s animal model. Brain Res Rev. 2006;52:275-292.
- Colaco CA, Ledesma MD, Harrington CR, Avila J. The role of the Maillard reaction in other pathologies: Alzheimer's disease. Nephrol Dial Transplant. 1996;11(Suppl 5):7-12.
pubmed - Janus C, Westaway D. Transgenic mouse models of Alzheimer's disease. Physiol Behav. 2001;73(5):873-886.
pubmed - Shi Q, Gibson GE. Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21(4):276-291.
pubmed - Ghribi O, DeWitt DA, Forbes MS, Arad A, Herman MM, Savory J. Cyclosporin A inhibits Al-induced cytochrome c release from mitochondria in aged rabbits. J Alzheimers Dis. 2001;3(4):387-391.
pubmed - Ghribi O, Herman MM, DeWitt DA, Forbes MS, Savory J. Abeta(1-42) and aluminum induce stress in the endoplasmic reticulum in rabbit hippocampus, involving nuclear translocation of gadd 153 and NF-kappaB. Brain Res Mol Brain Res. 2001;96(1-2):30-38.
pubmed - Ghribi O, Herman MM, Forbes MS, DeWitt DA, Savory J. GDNF protects against aluminum-induced apoptosis in rabbits by upregulating Bcl-2 and Bcl-XL and inhibiting mitochondrial Bax translocation. Neurobiol Dis. 2001;8(5):764-773.
pubmed - Ghribi O, DeWitt DA, Forbes MS, Herman MM, Savory J. Co-involvement of mitochondria and endoplasmic reticulum in regulation of apoptosis: changes in cytochrome c, Bcl-2 and Bax in the hippocampus of aluminum-treated rabbits. Brain Res. 2001;903(1-2):66-73.
pubmed - Ghribi O, Herman MM, Savory J. The endoplasmic reticulum is the main site for caspase-3 activation following aluminum-induced neurotoxicity in rabbit hippocampus. Neurosci Lett. 2002;324(3):217-221.
pubmed - Rao KSJ, Anitha S, Latha KS. Aluminium induced neurodegeneration in hippocampus of aged rabbits mimics AD. ALZ Rep. 2000;3:83-88.
pubmed - Savory J, Jagannatha KS Rao, Yue H, Philip R, Ledata, mary M Herman. Age-related hippocampal changes in Bcl3-2: Bax Ratio, Oxidative Stress, Redox-Active iron and apoptosis associated with Al induced neurodegeneration: Increased susceptibility with aging. Neurotoxicology. 1999;20:805-818.
- Savory J, Ghribi O, Forbes MS, Herman MM. Aluminium and neuronal cell injury: inter-relationships between neurofilamentous arrays and apoptosis. J Inorg Biochem. 2001;87:1-2.15-19.
pubmed - Savory J, Herman MM, Ghribi O. Intracellular mechanisms underlying aluminum-induced apoptosis in rabbit brain. J Inorg Biochem. 2003;97(1):151-154.
pubmed - Gackowski D, Rozalski R, Siomek A, Dziaman T, Nicpon K, Klimarczyk M, Araszkiewicz A,
et al . Oxidative stress and oxidative DNA damage is characteristic for mixed Alzheimer disease/vascular dementia. J Neurol Sci. 2008;266(1-2):57-62.
pubmed - Lovell MA, Markesbery WR. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer's disease. Nucleic Acids Res. 2007;35(22):7497-7504.
pubmed - Martin LJ. DNA damage and repair: relevance to mechanisms of neurodegeneration. J Neuropathol Exp Neurol. 2008;67(5):377-387.
pubmed - Moreira PI, Nunomura A, Nakamura M, Takeda A, Shenk JC, Aliev G, Smith MA,
et al . Nucleic acid oxidation in Alzheimer disease. Free Radic Biol Med. 2008;44(8):1493-1505.
pubmed - Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J. Increased levels of DNA breaks in cerebral cortex of Alzheimer's disease patients. Neurobiol Aging. 1990;11(3):169-173.
pubmed - Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J,
et al . Alzheimer's disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28(1):3-9.
pubmed - Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71(5):2034-2040.
pubmed - Lee SH, Kim I, Chung BC. Increased urinary level of oxidized nucleosides in patients with mild-to-moderate Alzheimer's disease. Clin Biochem. 2007;40:13-14.936-938.
pubmed - Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93(4):953-962.
pubmed - Francisconi S, Codenotti M, Ferrari-Toninelli G, Uberti D, Memo M. Preservation of DNA integrity and neuronal degeneration. Brain Res Brain Res Rev. 2005;48(2):347-351.
pubmed - Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35(16):5545-5555.
pubmed - Jacobsen E, Beach T, Shen Y, Li R, Chang Y. Deficiency of the Mre11 DNA repair complex in Alzheimer's disease brains. Brain Res Mol Brain Res. 2004;128(1):1-7.
pubmed - Tsujimura A, Matsuki M, Takao K, Yamanishi K, Miyakawa T, Hashimoto-Gotoh T. Mice Lacking the kf-1 Gene Exhibit Increased Anxiety- but not Despair-Like Behavior. Front Behav Neurosci. 2008;2:4.
pubmed - Johnson GV, Bailey CD. Tau, where are we now? J Alzheimers Dis. 2002;4(5):375-398.
pubmed - Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, Titani K,
et al . Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16(3):365-371, discussion 371-380.
pubmed - Chatterjee S, Sang TK, Lawless GM, Jackson GR. Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum Mol Genet. 2009;18(1):164-177.
pubmed - McLarnon JG, Ryu JK. Relevance of abeta1-42 intrahippocampal injection as an animal model of inflamed Alzheimer's disease brain. Curr Alzheimer Res. 2008;5(5):475-480.
pubmed - Emil Adamec, Vonsatte Jean Paul, Nixon Ralph A. DNA strand breaks in AD. Brain Res. 1999;849:67-77.
- Finneagan MM, Rettig S, Orvig CA. A Neutral water soluble Al complex of neurological interest. J Am Chem Soc. 1986;108:5033-5035.
- Savory J, Huang Y, Herman MM, Reyes MR, Wills MR. Tau immunoreactivity associated with aluminum maltolate-induced neurofibrillary degeneration in rabbits. Brain Res. 1995;669(2):325-329.
pubmed - Strazielle C, Dumont M, Fukuchi K, Lalonde R. Transgenic mice expressing the human C99 terminal fragment of betaAPP: effects on cytochrome oxidase activity in skeletal muscle and brain. J Chem Neuroanat. 2004;27(4):237-246.
pubmed - Shek JW, Wen GY, Wisniewski HM. Atlas of the rabbit brain & Spinal cord, Basel, Karger, (1986)..
- Hegde ML, Gupta VB, Anitha M, Harikrishna T, Shankar SK, Muthane U, Subba Rao,
et al . Studies on genomic DNA topology and stability in brain regions of Parkinson's disease. Arch Biochem Biophys. 2006;449:1-2.143-156.
pubmed - Scatchard G. The attraction of proteins for small molecules & ions. Ann NY Aca. Sci. US. 1949;51:660-672.
- Martin RB. The chemistry of aluminum as related to biology and medicine. Clin Chem. 1986;32(10):1797-1806.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.