










| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website http://www.jocmr.org |
Original Article
Volume 10, Number 8, August 2018, pages 636-640
The Characteristics and Trends in Adrenocortical Carcinoma: A United States Population Based Study
Eliza Sharmaa, Suyash Dahalb, Pratibha Sharmaa, Abani Bhandarib, Vishal Guptab, Birendra Amgaic, Sumit Dahalc, d
aDepartment of Internal Medicine, Maimonides Medical Center, Brooklyn, NY, USA
bDepartment of Medicine, KIST Medical College and Teaching Hospital, Lalitpur, Nepal
cDepartment of Medicine, Interfaith Medical Center, Brooklyn, NY, USA
dCorresponding Author: Sumit Dahal, Department of Medicine, Interfaith Medical Center, 1545 Atlantic Avenue, Brooklyn, NY 11213, USA
Manuscript submitted June 3, 2018, accepted June 20, 2018
Short title: Adrenocortical Carcinoma in US
doi: https://doi.org/10.14740/jocmr3503w
| Abstract | ▴Top |
Background: Adrenocortical carcinoma (ACC) is a rare malignancy with poor prognosis. Data on the incidence of ACC, however, are scarce and not recent. The purpose of this study was to characterize the tumor and the patients developing ACC over the last four decades using a large population based database.
Methods: We identified all cases of ACC diagnosed between 1973 - 2014 from the Surveillance, Epidemiology, and End Results-18 registry. Descriptive analyses were used for all extracted demographic, clinical, pathological, therapeutic and survival data, and were compared between the four time periods of 1973 to 1984, 1985 to 1994, 1995 to 2004 and 2005 to 2014 using Chi-square tests for categorical variables and one-way analysis of variance for continuous variables.
Results: There were a total of 2,014 cases of ACC between 1973 and 2014 with an age-adjusted incidence of 1.02 per million populations. The median age at diagnosis was 55 years with the majority of them being females and whites. The proportion of cases by different genders, races and age at diagnosis had not changed significantly over time. These malignancies were mostly the only primary malignancy, unilateral and of high grades at diagnosis. Surgical resection of the tumor remained the mainstay of treatment. However, there was a significant increase in the use of adjuvant radiotherapy, adjuvant chemotherapy and chemotherapy alone in recent times. The median survival time was 17 months, but continues to decrease in recent time periods.
Conclusions: ACC continues to be a rare malignancy in the United States. However, most cases continue to be diagnosed only in advanced stages and are associated with poor survival. These findings underline the need for specific diagnostics tools with new and more effective treatment options.
Keywords: Adrenocortical carcinoma; Adrenal malignancy; SEER database
| Introduction | ▴Top |
Adrenocortical carcinoma (ACC) is a rare malignancy with poor prognosis. The reported incidence is 0.5 - 2 cases per million populations per year [1, 2]. ACC may account for up to 14% of adrenal incidentalomas, and is one of the most common indications for adrenalectomy in an adrenal incidentaloma [3, 4]. Most ACCs occur as sporadic tumors but can rarely be associated with hereditary syndromes like Li-Fraumeni syndrome and Beckwith-Wiedemann syndrome [5]. It is more common in females and can occur at any age with a peak in fifth and sixth decades of life [2]. Data on the incidence of ACC, however, are scarce and not recent. The purpose of this study was to characterize the tumor and the patients developing ACC over the last four decades using a large population based database.
| Methods | ▴Top |
We made use of National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) data from 18 registries. This program has collected demographic, clinical, pathological, therapeutic and survival data on patients with ACC in the United States since 1973, and covers approximately 27.8% of United States population. We identified all cases of ACC diagnosed between 1973 and 2014 from the SEER 18 registry using the International Classification of Disease for Oncology, 3rd edition (ICD-O-3) code 8370/3. We excluded cases diagnosed at autopsy and those lost to follow-up.
The specific variables studied included patient demographics like year of diagnosis, gender, age and race; clinical and pathological data like the grade, laterality and sequence of the malignancy; and therapeutic and survival data like the treatment modalities, cause-specific mortality and survival period. Descriptive analyses were used for all extracted data. We used frequency session and case listing session of the SEER Stat software (version 8.3.5; March 6, 2018) to calculate the different frequencies and trends. The age-adjusted incidences of ACC were determined in the rate session of the SEER Stat software using the 2000 US standard population. Furthermore, for the cases diagnosed between 2000 and 2014, the survival session of the software was used to compare the observed survival of patients with ACC with the expected survival matched for age, sex, race, and year of diagnosis. Expected survival represented survival for a comparable group of individuals free of cancer, and was generated from expected survival life tables accompanying the SEER database.
To further analyze the trend in demographics and characteristics of ACC between 1973 and 2014, the time period was divided into four groups based on the year of diagnosis: 1973 to 1984, 1985 to 1994, 1995 to 2004 and 2005 to 2014. The different characteristics between these time groups were then compared using Chi-square tests for categorical variables and one-way analysis of variance (ANOVA) for continuous variables. All the calculations were done using SEER Stat software (version 8.3.5; March 6, 2018) and statistical package for social sciences (version 22).
| Results | ▴Top |
There were a total of 2,014 cases of ACC between 1973 and 2014. There has been a 977% increase in the number of cases between this time periods with an annual percentage change (APC) of 5.7% (Table 1). Nearly three-fifths (57%) of the cases were females with an APC of 6.2% as compared to 5% for males. The median age at diagnosis was 55 years, and more than 60% of the cases were diagnosed between 40 to 70 years of age. Most of the cases (86%) were seen in White Americans. The proportion of cases by different genders, races and age at diagnosis had not changed significantly over the four time periods (Table 2).
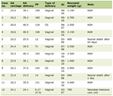 Click to view | Table 1. Overall Characteristics of Adrenocortical Carcinoma Between 1973 and 2014 |
Click to view | Table 2. Trend in the Characteristics of Adrenocortical Carcinoma |
The overall age-adjusted incidence of ACC was 1.02 per 1,000,000 populations, and did not change significantly between 1973 and 2014 (Fig. 1). Almost one-fifth of the cases of ACC were associated with other primary malignancy. These cases were predominantly unilateral, affecting the left adrenal gland more commonly than the right (51.6% vs. 43.4%). Most of the carcinomas were of high grades at diagnosis. Nearly two-thirds of the cases were either poorly differentiated (grade III) or undifferentiated (grade IV) at diagnosis, and this proportion has only increased in the four time periods (42% vs. 64.7% vs. 64.6% vs. 71%, P = 0.04). Surgical resection of the tumor remains the mainstay of treatment of ACC. However, less than three-fourths of the patients underwent surgical resection with one-tenth of them also receiving adjuvant radiotherapy and one-fourth receiving adjuvant chemotherapy. While the proportion of patients undergoing surgical resection has remained unchanged between 1973 and 2014, there has been a significant increase in the use of adjuvant radiotherapy, adjuvant chemotherapy and chemotherapy alone. The overall cause-specific mortality rate was 52% with a median survival time of 17 months. For the cases undergoing surgical resection of the tumor, the median survival time was much higher at 28 months. The observed survival of patients with ACC diagnosed between 2000 and 2014 was well below the expected survival matched for age, sex, race, and year of diagnosis (Fig. 2). Alarmingly, the mean survival time has been on a downward trend in the last two of the four time periods (77.35 ± 132.7 vs. 80.82 ± 105.7 vs. 64.04 ± 69.79 vs. 25.76 ± 29.184, P < 0.05).
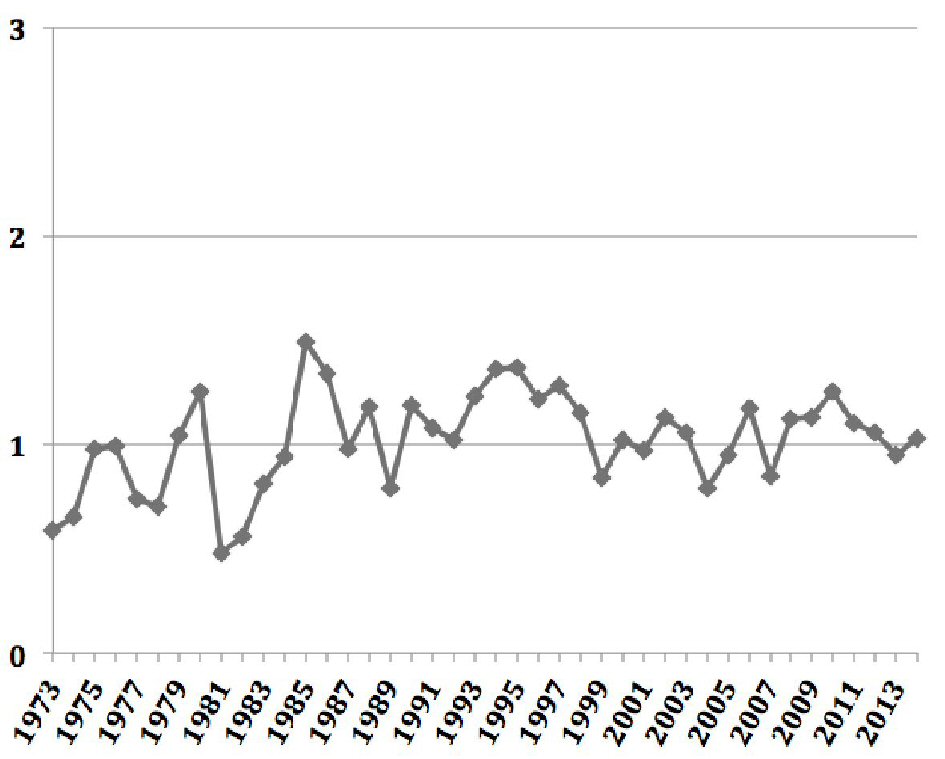 Click for large image | Figure 1. Age-adjusted annual incidence of adrenocortical carcinoma over time. |
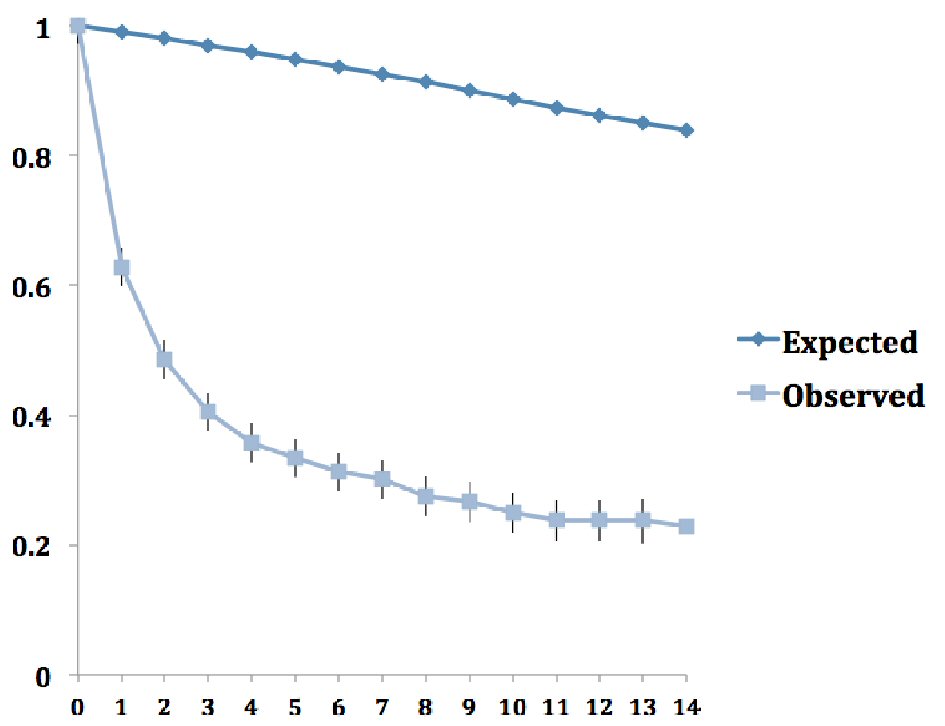 Click for large image | Figure 2. Observed and expected survival rates for adrenocortical carcinoma over time. |
| Discussion | ▴Top |
ACC is a rare malignancy. The annual incidence in our study was found to be 1.02 per 1,000,000 populations. This is consistent with previously reported incidence of 0.5 - 2 cases per million populations per year [1, 2]. Kebebew et al reported an annual incidence of 0.72 per million in the United States between 1973 and 2000. The slightly higher incidence in our study compared to the findings by Kebebew et al may be due to the increased use of more sensitive imaging studies in recent years that incidentally identify ACC [3]. ACC may in fact account for up to 14% of adrenal incidentalomas, and is one of the most common indications for adrenalectomy in adrenal incidentaloma [3, 4, 6]. While this present study and the prior one by Kebebew et al show the annual incidence of ACC to be largely unchanged in the United States, there seems to a trend towards a declining annual incidence in Netherlands [7]. The reasons for these findings, however, remain speculative.
There was predominance of females and whites with a female to male ratio of 1.34:1 and Whites to non-Whites ratio of 6.3:1; and these were on par with previous studies [2, 7]. Historically, a bimodal age distribution has been described for ACC, with one peak in early childhood and another peak in fourth and fifth decades of life [8, 9]. Our study had no such bimodal age distribution, and a single peak was seen in the sixth and seventh decades of life with the median age at diagnosis of 55 years. Few previous studies reported a similar absence of bimodal age distribution with a single peak in sixth decade of life [2, 7]. The mean age at diagnosis has remained unchanged over these years.
Most cases of ACC occur sporadically. ACC was the only primary malignancy in 80% of our cases. However, the rest one-fifth of our patients had at least one other malignancy. A part of this high proportion could be explained by the fact that ACC can also occur in association with hereditary syndromes like Li-Fraumeni syndrome, Beckwith-Wiedemann syndrome and multiple endocrine neoplasia type I [5, 10]. About 95% of the cases of ACC involved a unilateral adrenal gland, with the left gland slightly more affected than the right one. This peculiar difference in the lateralization was also seen in a previous study [7]. However, the explanation for this remains elusive. Patients may present with symptoms related to either the mass effect of the tumor or hormonal overproduction. However, a significant number of patients are asymptomatic and only discovered incidentally on imaging studies for non-adrenal symptoms [3]. Majority of the tumors was of high grade at diagnosis and their proportion has been increasing with time. While it is expected that more ACCs would be discovered at earlier stages of the disease due to the increased use of sensitive imaging studies in recent years with subsequent improvement in survival, multiple studies have shown findings to the contrary [2, 7, 11-13]. Kebebew et al and Kerkhofs et al reported no changes in the trend of the ACC cases being diagnosed mostly in advanced stages of the disease [2, 7]. One possible explanation is that due to limitations in the specificity of current diagnostic tools, small ACCs may be initially misclassified as benign and are only picked up later as ACC in their advanced stages. This, compounded by the lack of new, effective therapies, may translate to non-improving or worsening survival in recent time periods as seen in our study or the one by Kerkhofs et al. Like in our study, surgical resection of the primary tumor remains the mainstay of therapy, and is often curative for local disease and may yield survival benefits in advanced diseases [14, 15]. Adrenolytic agent mitotane is mainly used for metastatic diseases, with the addition of cytotoxic chemotherapy for more advanced stages [16, 17]. The use of adjuvant radiotherapy and adjuvant mitotane, previously reported to increase the recurrence-free survival, was found to be increasing in recent years in our study [18, 19].
In conclusion, ACC continues to be a rare malignancy in the United States with an annual incidence of 1.02 per million populations. However, most cases continue to be diagnosed only in advanced stages and are associated with poor survival. These findings underline the need for specific diagnostics tools with new and more effective treatment for adrenocortical cancers.
Acknowledgments
This study utilized the SEER 18 database. The interpretation of this data is the sole responsibility of the authors. The authors acknowledge the efforts of Surveillance Research Program, National Cancer Institute and the SEER program tumor registries in the creation of the SEER database.
Conflict of Interest
None.
| References | ▴Top |
- Wooten MD, King DK. Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer. 1993;72(11):3145-3155.
doi - Kebebew E, Reiff E, Duh QY, Clark OH, McMillan A. Extent of disease at presentation and outcome for adrenocortical carcinoma: have we made progress? World J Surg. 2006;30(5):872-878.
doi pubmed - Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev. 2004;25(2):309-340.
doi pubmed - Ambrosi B, Peverelli S, Passini E, Re T, Ferrario R, Colombo P, Sartorio A, et al. Abnormalities of endocrine function in patients with clinically "silent" adrenal masses. Eur J Endocrinol. 1995;132(4):422-428.
doi pubmed - Lerario AM, Moraitis A, Hammer GD. Genetics and epigenetics of adrenocortical tumors. Mol Cell Endocrinol. 2014;386(1-2):67-84.
doi pubmed - Sturgeon C, Kebebew E. Laparoscopic adrenalectomy for malignancy. Surg Clin North Am. 2004;84(3):755-774.
doi pubmed - Kerkhofs TM, Verhoeven RH, Van der Zwan JM, Dieleman J, Kerstens MN, Links TP, Van de Poll-Franse LV, et al. Adrenocortical carcinoma: a population-based study on incidence and survival in the Netherlands since 1993. Eur J Cancer. 2013;49(11):2579-2586.
doi pubmed - Ng L, Libertino JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol. 2003;169(1):5-11.
doi - Wajchenberg BL, Albergaria Pereira MA, Medonca BB, Latronico AC, Campos Carneiro P, Alves VA, Zerbini MC, et al. Adrenocortical carcinoma: clinical and laboratory observations. Cancer. 2000;88(4):711-736.
doi - Gatta-Cherifi B, Chabre O, Murat A, Niccoli P, Cardot-Bauters C, Rohmer V, Young J, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d'etude des Tumeurs Endocrines database. Eur J Endocrinol. 2012;166(2):269-279.
doi pubmed - Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91(6):2027-2037.
doi pubmed - Icard P, Goudet P, Charpenay C, Andreassian B, Carnaille B, Chapuis Y, Cougard P, et al. Adrenocortical carcinomas: surgical trends and results of a 253-patient series from the French Association of Endocrine Surgeons study group. World J Surg. 2001;25(7):891-897.
doi pubmed - Chen DY, Sosa RE, Scherr DS. Treatment of adrenocortical carcinoma: contemporary outcomes. Curr Urol Rep. 2004;5(1):65-72.
doi pubmed - Berruti A, Baudin E, Gelderblom H, Haak HR, Porpiglia F, Fassnacht M, Pentheroudakis G, et al. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii131-138.
doi pubmed - Erdogan I, Deutschbein T, Jurowich C, Kroiss M, Ronchi C, Quinkler M, Waldmann J, et al. The role of surgery in the management of recurrent adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98(1):181-191.
doi pubmed - Haak HR, Hermans J, van de Velde CJ, Lentjes EG, Goslings BM, Fleuren GJ, Krans HM. Optimal treatment of adrenocortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br J Cancer. 1994;69(5):947-951.
doi pubmed - Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012;366(23):2189-2197.
doi pubmed - Terzolo M, Ardito A, Zaggia B, Laino F, Germano A, De Francia S, Daffara F, et al. Management of adjuvant mitotane therapy following resection of adrenal cancer. Endocrine. 2012;42(3):521-525.
doi pubmed - Berruti A, Fassnacht M, Baudin E, Hammer G, Haak H, Leboulleux S, Skogseid B, et al. Adjuvant therapy in patients with adrenocortical carcinoma: a position of an international panel. J Clin Oncol. 2010;28(23):e401-402; author reply e403.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.